

 下载:
下载:

-
创伤性脑损伤(Traumatic Brain Injury, TBI)是指由外力作用导致脑组织结构受损或功能障碍的神经系统疾病[1-2]。作为全球面临的重大公共卫生问题,TBI每年新增病例约
2700 万至6900 万例,其中中重度患者的死亡率高达三分之一。据统计,中国每年新增TBI病例超400万例,高居全球首位[3]。最新全球疾病负担研究指出,2020年以来我国TBI发病率呈反弹上升趋势,临床救治面临更加严峻的考验[4]。TBI的继发性损伤是一个多因素驱动的渐进过程,常伴随严重的线粒体功能障碍与能量耗竭。它通常发生在伤后数小时至数天,由一系列相互交织的病理生理和分子瀑布效应所引发。这种进行性的演变为药物干预提供了宝贵的“时间窗口”[5]。然而,由于其病理机制错综复杂,目前的临床干预多局限于控制颅内压、缓解脑水肿等对症支持治疗,尚缺乏能够直接阻断神经元死亡进程的特异性靶向药物[6]。
在众多的潜在干预靶点中,聚ADP核糖聚合酶(Poly(ADP-ribose)polymerase,PARP)的过度激活被认为是引发TBI后能量代谢危机的关键因素[7]。研究表明,TBI诱导的DNA损伤会造成PARP过度激活,导致细胞内NAD+和ATP被大量消耗,从而引发神经元的凋亡与坏死。然而,传统的全身性PARP抑制剂在阻断该级联反应的同时,往往会因过度抑制正常PARP的生理功能而产生严重的神经毒副作用。值得注意的是,Szczesny等的研究揭示了PARP的功能表现出显著的亚细胞定位依赖性:与参与细胞核DNA修复的核内PARP不同,位于线粒体内的PARP会抑制线粒体DNA的修复并加剧能量耗竭[8]。因此,构建线粒体精准靶向的PARP抑制剂递送系统,在特异性抑制线粒体PARP异常激活的同时,保留细胞核正常的生理修复功能,已成为阻断神经元死亡的潜在治疗策略。
基于此,本研究选取临床一线PARP抑制剂奥拉帕尼(Olaparib,OLA)作为模型药物[9-10]。针对OLA原药水溶性差且缺乏特定亚细胞靶向性的临床应用瓶颈,设计并合成了一种具有线粒体靶向功能的新型化合物TPP-OLA。该化合物在保留OLA核心药效基团的基础上,引入了膜电位驱动的线粒体靶向基团三苯基膦 [11-13]。在确证其化学结构与体外酶抑制活性后,本研究将其进一步包载于脂质体中以改善整体成药性。
为实现对病灶的精准干预,本研究构建了表面修饰有狂犬病毒糖蛋白衍生物(DSPE-PEG2000-RVG29)的脑靶向脂质体系统(TPP-OLA/Lip)[14-15]。该系统融合了长循环与多级靶向的设计理念:理论上,RVG29肽不仅能特异性识别血脑屏障内皮细胞表面的烟碱型乙酰胆碱受体,通过跨胞吞作用介导脂质体穿透血脑屏障,还能进一步促进中枢神经元对载药系统的靶向摄取;进入细胞后,结合TPP-OLA分子自带的TPP阳离子基团,系统可实现受损神经元内部的线粒体特异性富集。该载药平台有望完成“跨越血脑屏障-神经元摄取-线粒体靶向”的级联递送,旨在从制剂学维度为 TBI 的神经保护治疗提供物质基础与实验依据。
-
Isolera One-自动快速制备色谱仪(瑞典 Biotage 公司);Quantum-I 400-核磁共振波谱仪(武汉中科牛津波谱技术有限公司);Agilent
6230 TOF LC/MS-液相色谱-飞行时间质谱仪(美国 Agilent Technologies 公司);Arc HPLC-高效液相色谱仪(美国 Waters 公司);AR224CN-电子天平(美国 Ohaus 公司);OSB-2200 -旋转蒸发仪(日本 EYELA 公司);SHZ-D(III)-循环水式真空泵(巩义市予华仪器有限公司);DZF-6020 -真空干燥箱(上海一恒科学仪器有限公司);SCIENTZ-IID-超声波细胞粉碎机(宁波新芝生物科技股份有限公司);Litesizer 500-纳米粒度及电位分析仪(奥地利 Anton Paar 公司);Turbiscan Tower-多重光散射仪(法国 Formulaction 公司);HT7800-透射电子显微镜(日本 Hitachi 公司);Spark-多功能酶标仪(瑞士 Tecan 公司);CF1524R-高速冷冻离心机(美国 SCILOGEX 公司)。 -
5-[(3,4-二氢-4-氧代-1-酞嗪基)甲基]-2-氟苯甲酸、哌嗪-1-羧酸叔丁酯(上海毕得医药科技股份有限公司);3-溴丙酰氯(北京沃凯生物科技有限公司);三苯基膦(北京伊诺凯科技有限公司);TBTU、HATU(上海阿拉丁生化科技股份有限公司);大豆磷脂(CS-95)、胆固醇、DSPE-mPEG2000(上海艾伟拓医药科技有限公司);DSPE-PEG2000-RVG29(广州市碳水生物科技有限公司);PARP比色法检测试剂盒(美国 R&D Systems公司);2% 磷钨酸负染色液、10% Triton X-100 水溶液(北京索莱宝科技有限公司);PBS缓冲液(武汉赛维尔生物科技有限公司);色谱纯甲醇、乙腈(美国 Thermo Fisher 公司);核磁共振氘代溶剂CDCl3、DMSO-d6(上海安耐吉化学有限公司);Amicon Ultra 0.5 ml超滤管(30K MWCO,美国 Millipore 公司)。实验中所用其余化学试剂及溶剂均为市售分析纯。
-
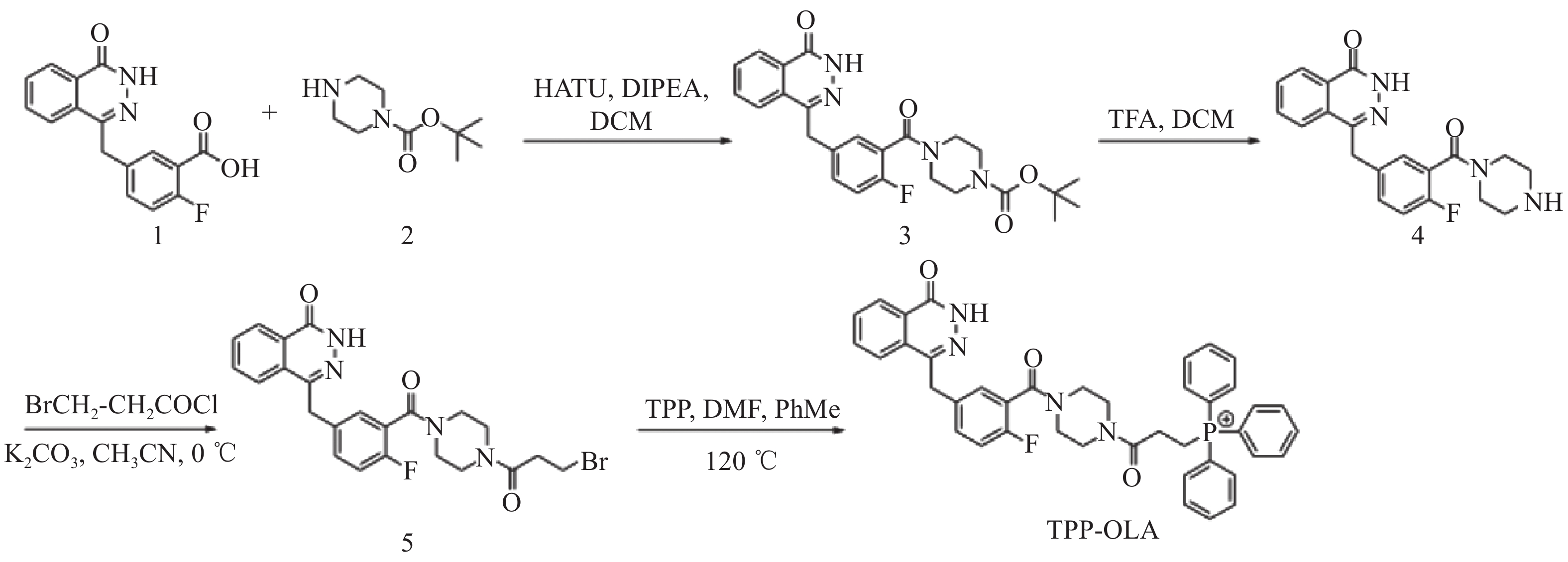
以5-[(3,4-二氢-4-氧代-1-酞嗪基)甲基]-2-氟苯甲酸为起始原料,经酰胺化、脱保护、N-酰化及季铵化4步反应合成目标化合物TPP-OLA(图1)。
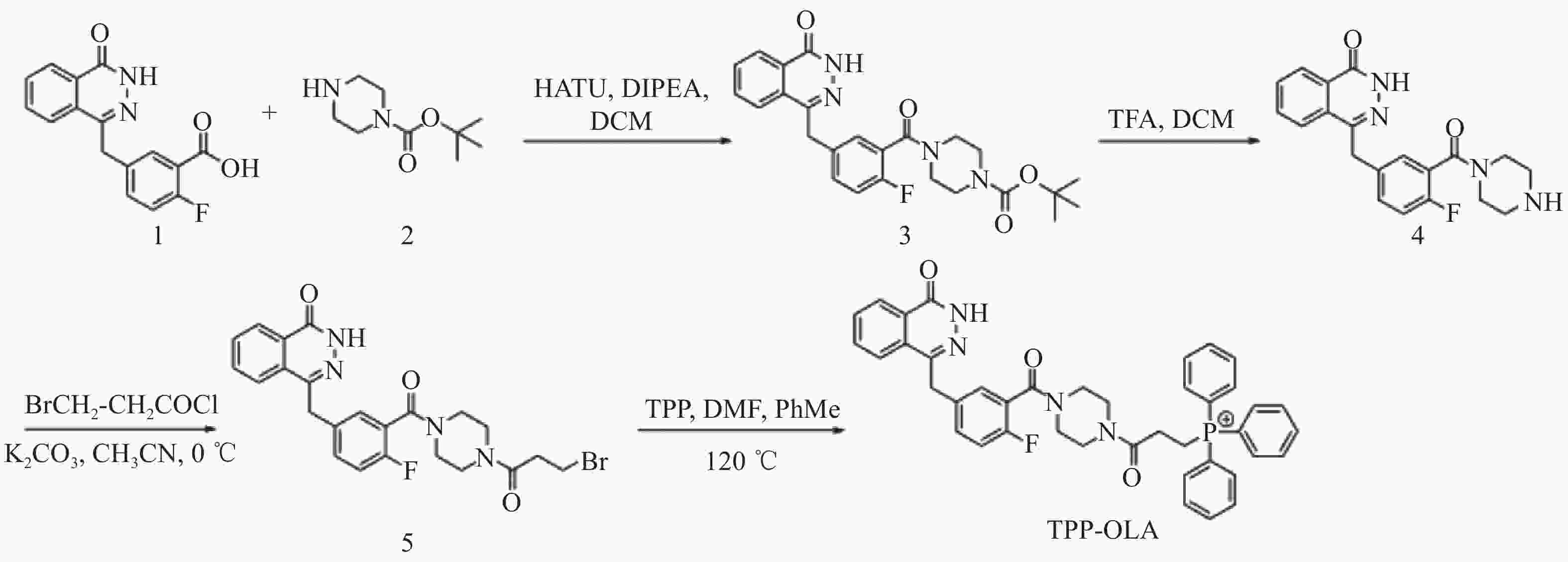
图 1 TPP-OLA的合成路线
-
将化合物1(5.00 g, 16.76 mmol)溶于50 ml二氯甲烷中,于冰浴下加入缩合剂HATU(8.29 g,21.79 mmol),搅拌10 min至完全溶解后,滴加N,N-二异丙基乙胺(5.85 g, 45.26 mmol),继续搅拌30 min。随后缓慢滴加化合物2(4.06 g, 21.79 mmol)的二氯甲烷溶液(20 ml),撤去冰浴,自然升至室温反应3 h。TLC(石油醚∶乙酸乙酯=1:2,v/v,含1%甲酸)监测至反应完全。反应结束后,将反应液用二氯甲烷稀释,依次用饱和碳酸氢钠水溶液、饱和食盐水和蒸馏水洗涤。有机相用无水硫酸钠干燥,过滤,减压浓缩得粗品。粗产物经快速制备色谱纯化,以石油醚-乙酸乙酯(含1%甲酸)为洗脱剂(极性比例从100∶0升至40∶60),收集目标组分并减压浓缩,得淡黄色固体中间体3(6.67 g,产率85.3%)。1H NMR(600 MHz, CDCl3)δ: 11.12(s, 1H), 8.52 – 8.41(m, 1H), 7.84 – 7.60(m, 3H), 7.36 – 7.28(m, 2H), 7.02(t, J = 8.8 Hz, 1H), 4.28(s, 2H), 3.73(s, 2H), 3.51(d, J = 9.0 Hz, 2H), 3.38(t, J = 5.2 Hz, 2H), 3.26(s, 2H), 1.46(s, 9H); ESI-MS m/z:489.18 [M+Na]+。
-
将中间体3(5.00 g,10.72 mmol)溶于50 ml二氯甲烷中,于0°C冰水浴中冷却。搅拌下缓慢滴加三氟乙酸(33 ml,428.71 mmol)。滴加完毕后,撤去冰浴,升至室温反应2 h。TLC(二氯甲烷∶甲醇=5∶1,v/v,含1% 氨水)监测至反应完全。将反应液减压浓缩,除去大部分溶剂及过量的三氟乙酸。残余物加适量蒸馏水稀释,冰浴下用饱和碳酸氢钠水溶液缓慢调节pH至8~9。用乙酸乙酯多次萃取,合并有机相,依次用饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩,得白色固体中间体4(3.51 g,产率89.3%)。1H NMR(600 MHz, DMSO-d6)δ: 12.61(s, 1H), 8.26(d, J = 7.8 Hz, 1H), 7.97 – 7.80(m, 3H), 7.45 – 7.18(m, 3H), 4.32(s, 2H), 3.59(br s, 2H), 3.14(br s, 2H), 2.78(t, J = 5.0 Hz, 2H), 2.64(br s, 2H); ESI-MS m/z: 367.15 [M+H]+。
-
将中间体4(5.00 g, 13.65 mmol)溶于无水乙腈(100 ml)中,于0 °C下加入无水碳酸钾(5.66 g, 40.94 mmol),搅拌成悬浮液。在氮气保护下,缓慢滴入3-溴丙酰氯(4.68 g, 27.29 mmol)的无水乙腈溶液(20 ml)。滴加完毕后,维持0 °C继续搅拌反应8 h。TLC(二氯甲烷∶甲醇=10∶1,v/v,含1% 氨水)监测至反应完全。反应液抽滤除去不溶性无机盐,滤饼用少量乙腈洗涤。合并滤液并减压浓缩。残余物溶于二氯甲烷,依次用蒸馏水和饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩。粗产物经快速制备色谱纯化,以二氯甲烷-甲醇(含1%氨水)为洗脱体系进行梯度洗脱(极性比例从100:0升至9:1),收集目标组分并减压浓缩,得淡黄色固体中间体5(5.10 g,产率74.6%)。1H NMR(600 MHz, DMSO-d6)δ: 12.59(s, 1H), 8.26(d, J = 7.9 Hz, 1H), 8.03 – 7.76(m, 3H), 7.49 – 7.18(m, 3H), 4.33(s, 2H), 3.75 – 3.46(m, 6H), 3.42 – 3.34(m, 2H), 3.22 – 3.14(m, 2H), 3.03 – 2.90(m, 2H); ESI-MS m/z: 523.06 [M+Na]+。
-
称取中间体5(2.00 g, 3.99 mmol)和三苯基膦(4.19 g, 15.96 mmol)置于250 ml圆底烧瓶中。体系抽真空并充入氮气后,加入10 ml DMF搅拌溶解,随后加入100 ml甲苯。将反应液升温至120 °C,回流反应24 h。TLC(二氯甲烷∶甲醇=10∶1,v/v,含1%氨水)监测至反应不再转化。反应结束后自然冷却至室温,减压旋蒸除去大部分溶剂,得黏稠状残余物。向残余物中加入蒸馏水与乙酸乙酯,剧烈振摇后静置分层,弃去乙酸乙酯相。水相用二氯甲烷多次萃取,合并二氯甲烷相,无水硫酸钠干燥。过滤,减压浓缩得粗产物。粗产物经快速制备色谱纯化,以二氯甲烷-甲醇为洗脱体系进行梯度洗脱(极性比例从100:1升至1:1),收集目标组分并减压浓缩,得白色粉末状目标化合物TPP-OLA(1.03 g,产率 33.7%)。1H NMR(600 MHz, DMSO-d6)δ: 12.60(s, 1H), 8.25-8.22(m, 1H), 7.96-7.73(m, 18H), 7.45(ddd, J = 8.0, 5.0, 2.2 Hz, 1H), 7.35-7.30(m, 1H), 7.23(dt, J = 9.3, 4.7 Hz, 1H), 4.32(d, J = 4.5 Hz, 2H), 3.81-3.72(m, 2H), 3.59-3.47(m, 2H), 3.45-3.34(m, 2H), 3.25(dt, J = 15.6, 5.2 Hz, 2H), 3.07(d, J = 6.6 Hz, 2H), 2.84-2.70(m, 2H); 13C NMR(151 MHz, DMSO-d6)δ: 167.67, 167.58, 167.49, 164.04, 163.97, 159.37, 157.16, 155.54, 155.47, 144.82, 134.85, 134.83, 133.77, 133.71, 133.48, 131.85, 131.80, 131.54, 130.19, 130.10, 129.05, 128.86, 127.87, 126.04, 125.46, 123.48, 123.35, 118.82, 118.79, 118.25, 118.22, 116.01, 115.93, 115.86, 115.78, 54.92, 46.30, 45.92, 44.57, 44.05, 41.61, 41.29, 41.03, 40.97, 36.41, 25.54, 17.35, 17.26, 16.99, 16.90;31P NMR(243 MHz, DMSO-d6)δ: 25.05(s);19F NMR(565 MHz, DMSO-d6)δ: -119.72(s);ESI-HRMS m/z: 计算值C41H37FN4O3P+ [M]+
683.2582 ,测量值683.2582 。 -
本研究采用PARP活性检测试剂盒评价TPP-OLA的体外酶抑制作用。将预包被组蛋白的96孔板用1× PARP缓冲液水合后,依次加入梯度稀释的TPP-OLA或阳性对照奥拉帕尼(最终反应浓度为为10−6~1 µM),并加入PARP-HSA酶液。体系于室温孵育10 min后,加入1× PARP混合液启动反应,并于室温避光孵育60 min。反应结束后,依次经洗涤、加入Strep-HRP工作液及TACS-Sapphire底物进行显色。最后用5%磷酸溶液终止反应,使用酶标仪测定450 nm处的吸光度。以药物浓度的对数值为横坐标,相应的酶抑制率为纵坐标,利用GraphPad Prism软件进行非线性拟合,计算半数抑制浓度(IC50)。
-
采用薄膜水化法制备TPP-OLA脑靶向脂质体。精密称取大豆磷脂295 mg、胆固醇15 mg、DSPE-mPEG2000 20 mg、DSPE-PEG2000-RVG29 30 mg以及目标化合物 TPP-OLA 5 mg,共同置于100 ml茄形瓶中。加入6 ml三氯甲烷,超声使脂质材料与药物完全溶解。于50 °C水浴、120 r/min条件下,减压旋转蒸发除去有机溶剂,直至瓶壁形成一层均匀、透明的脂质薄膜。真空干燥过夜以彻底除去残留溶剂,加入5 ml高纯水作为水化介质,在50°C水浴、120 r/min旋转水化30 min,使薄膜脱落形成脂质混悬液。随后,采用探头超声仪(功率200 W,工作2 s,间歇2 s)在冰水浴下处理15 min,即得TPP-OLA/Lip胶体溶液。
-
精密量取100 μl TPP-OLA/Lip胶体溶液,使用高纯水稀释10倍后,置于专用样品池中,采用纳米粒度及电位分析仪测定其平均粒径、多分散系数(polydispersity index, PDI)以及Zeta电位。所有测量均在室温下平行进行3次,以考察体系的粒度分布、均一性及表面电荷特征。
-
采用超滤离心法结合HPLC测定TPP-OLA/Lip的包封率(encapsulation efficiency, EE%)。
游离药物量测定:精密量取500 μl TPP-OLA/Lip脂质体溶液,置于超滤离心管(截留分子量30 kDa)中,于4℃、
12600 r/min条件下离心15 min。收集底部外管中的澄清滤液,加入适量乙腈(调节溶剂比例至水∶乙腈 = 6∶4, v/v),转移至微量进样瓶中,即得游离药物供试品溶液。总药物量测定:精密量取100 μl TPP-OLA/Lip脂质体溶液,加入1.9 ml乙腈并剧烈涡旋振荡以彻底破乳。于4℃、
12600 r/min条件下离心15 min。精密吸取适量上清液,使用高纯水稀释(调节溶剂比例至水∶乙腈 = 6∶4, v/v),转移至微量进样瓶中,即为含总药物的供试品溶液。将上述两份供试品溶液按建立好的HPLC方法进行进样分析,根据标准曲线分别计算出游离药物质量(Wfree)和总药物质量(Wtotal)。包封率的计算公式如下:
$$ \text{EE\%} = \left(1-\frac{{W}_{\text{free}}}{{W}_{\text{total}}}\right)\times 100\text{%} $$ -
基于本课题组前期预实验确立的基础制备工艺,为进一步获得最优的载药制剂处方,本研究重点对影响靶向脂质体载药性能的关键变量进行了单因素考察。以脂质体的包封率、平均粒径、PDI及Zeta电位为评价指标。
药脂比的筛选:固定脂质骨架的总量及比例不变,分别以目标化合物TPP-OLA的投料量(1、2.5、5、7.5、10 mg)为自变量制备TPP-OLA/Lip脂质体。考察不同药脂比对制剂粒径、PDI、包封率及Zeta电位的影响,确定处方中药物与脂质材料的最佳投料比例。
铺膜溶剂的筛选:在确定最佳药物投料量的基础上,以铺膜所用有机溶剂的种类及体积组合(12 ml三氯甲烷、6 ml三氯甲烷、8 ml三氯甲烷 + 4 ml甲醇、4 ml三氯甲烷 + 2 ml甲醇)为自变量制备TPP-OLA/Lip脂质体。考察不同铺膜溶剂体系对薄膜形成状态以及最终脂质体粒径、PDI、包封率及Zeta电位的影响。
-
取适量新制备的TPP-OLA/Lip胶体溶液,使用高纯水稀释20倍。取微量稀释液滴加于铜网正面,静置10 min后,用滤纸从边缘吸去多余液体。随后,滴加2%磷钨酸负染液进行染色,约2 min后再次用滤纸吸干多余染液。待样品于室温下自然干燥后,置于透射电子显微镜(TEM)下观察脂质体的微观形态。
-
采用多重光散射技术对脂质体分散体系的物理稳定性进行评价。取适量新制备的 TPP-OLA/Lip置于专用的测量池中,采用多重光散射仪于室温下进行垂直动态扫描。设定扫描考察周期为6 d,通过仪器记录待测样品在考察周期内背散射光(ΔBS)和透射光(ΔT)的变化,以定量考察脂质体体系的稳定性。
-
采用动态透析法考察TPP-OLA/Lip的体外释药行为。将透析袋(截留分子量3 kDa)于蒸馏水中浸泡活化24 h后备用。精密量取适量TPP-OLA/Lip胶体溶液及同浓度的游离 TPP-OLA标准液,分别装入透析袋中,用透析夹严密封闭两端。
将装有样品的透析袋完全浸没于100 ml释放介质(含适量增溶剂的PBS,pH 7.4)中。置于恒温水浴摇床内,设定水浴温度为37℃,振摇转速为100 r/min。分别于设定的时间点(0.5、1、2、4、6、8、24、48和72 h)精密吸取释放液2.0 ml,并立即向体系中补加等体积且预热的空白释放介质。将收集的样品经HPLC仪进行检测,计算各时间点药物的累积释放率,绘制体外释药曲线。
-
按照“2.1”项下合成路线,成功制备得到目标化合物TPP-OLA及其各步中间体。其化学结构均已通过 1H-NMR、ESI-MS等波谱手段确证(详见2.1节),所得产物纯度符合后续脑靶向脂质体制备及体外评价的要求。
-
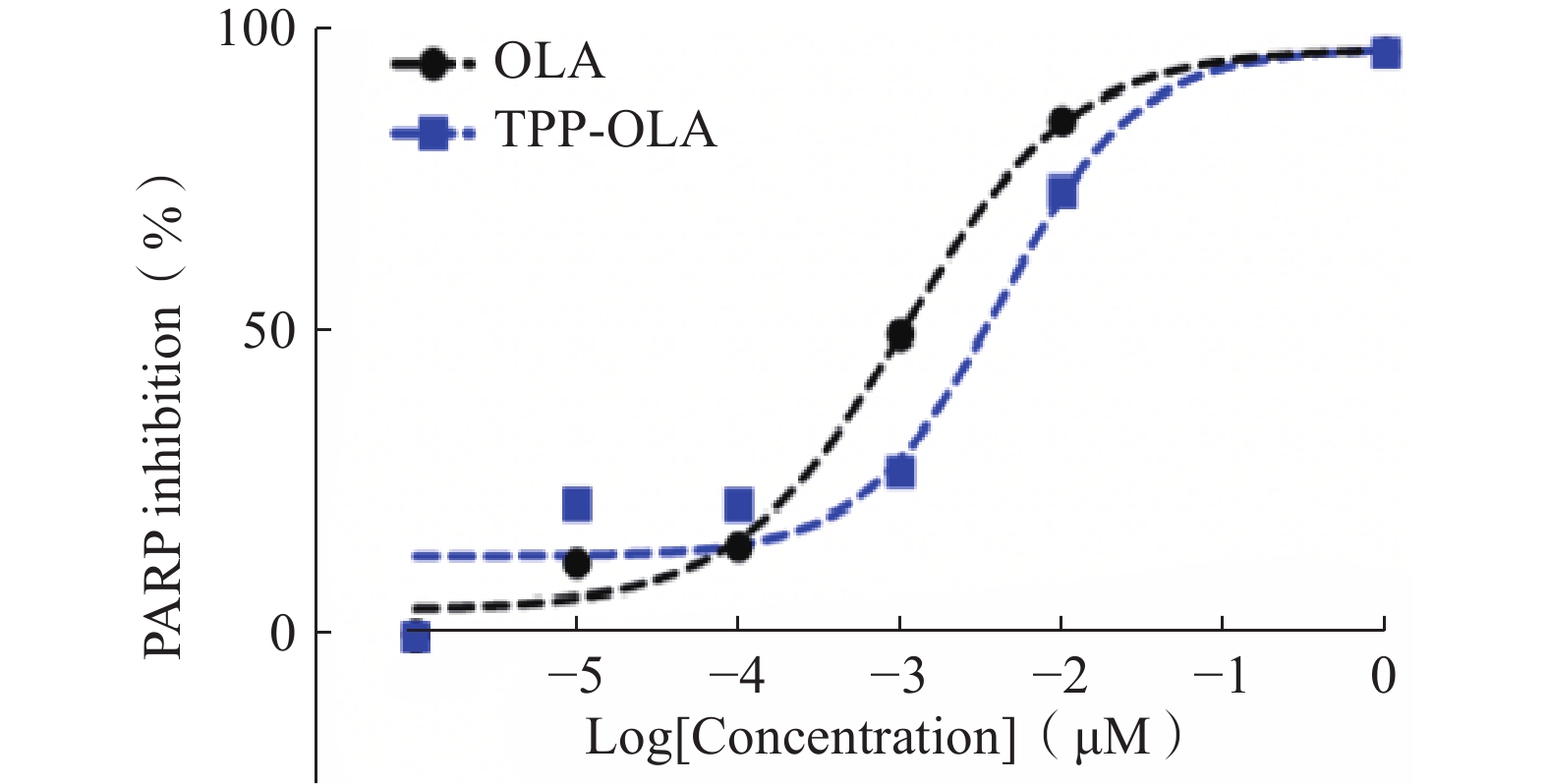
在10−6~1 μM的浓度范围内,TPP-OLA与母体药物OLA对PARP酶均呈现出显著的浓度依赖性抑制效应(图2)。经非线性曲线拟合求得,OLA的半数抑制浓度(IC50)为1.0 nM,而修饰后的TPP-OLA的IC50为4.1 nM。
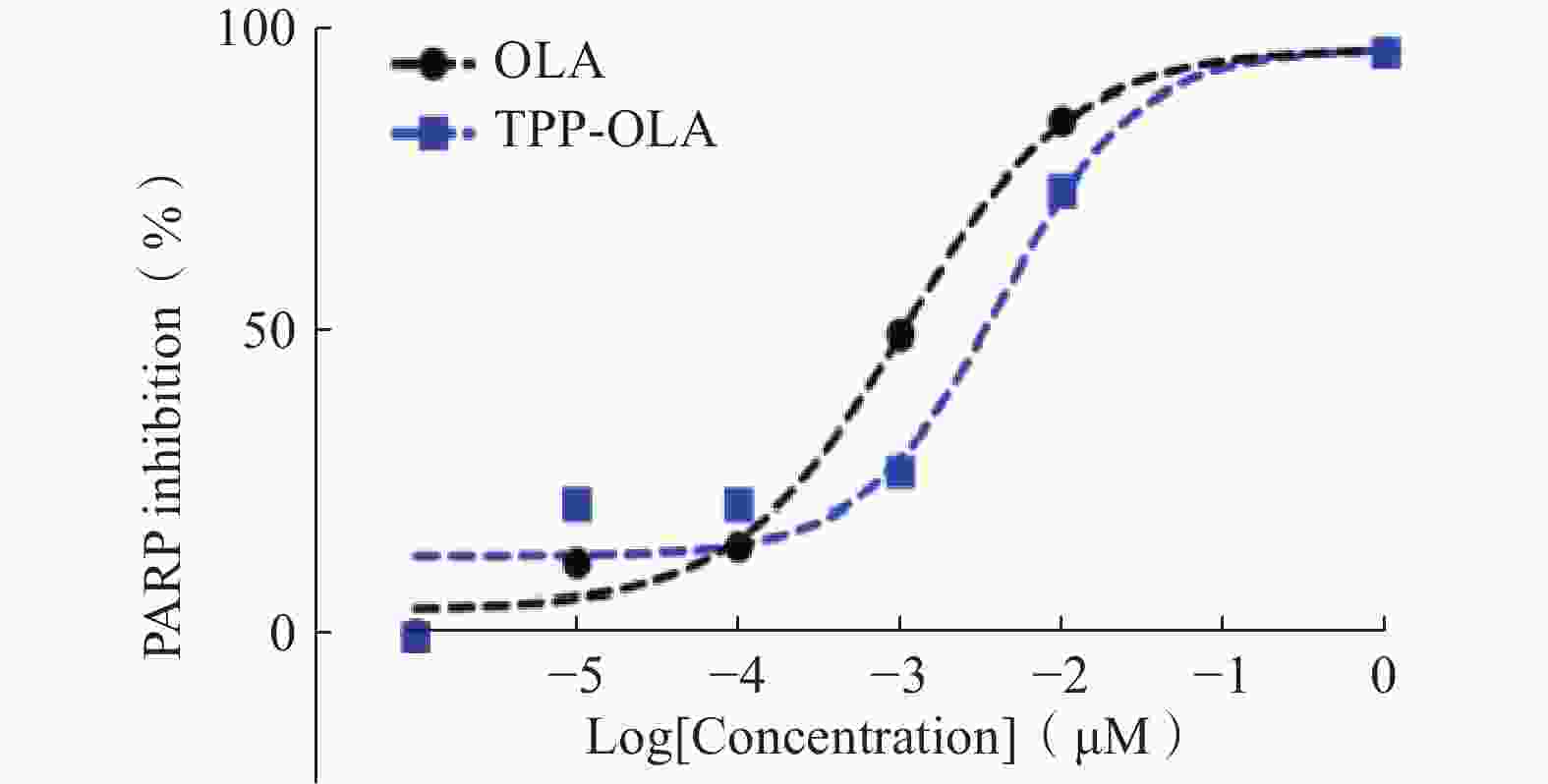
图 2 TPP-OLA与OLA对PARP酶活性的抑制曲线
结果表明,大体积的亲脂性阳离子(三苯基膦基团)及连接臂的引入增加了分子的空间位阻,导致其抑制活性较OLA略有降低。尽管如此,TPP-OLA的IC50仍维持在纳摩尔级别,且在给药浓度达到1 μM时,二者的酶抑制率基本持平(均接近100%),说明TPP-OLA依然具备强效的酶抑制活性。
综上所述,结构修饰后的TPP-OLA有效保留了原药的核心抑制功能,体外抑制效果符合预期。
-
采用Welch Ultimate ODS-3色谱柱(4.6 mm×150 mm,5 µm,月旭科技)。流动相采用流动相A(0.05 mol/l磷酸二氢钾溶液,含0.1% 三乙胺,调节pH至6.09)与流动相B(乙腈)等梯度洗脱(A∶B = 60∶40,v/v)。流速1.0 ml/min,柱温35℃,检测波长236 nm,进样量10 µl。样品处理时,将载药脂质体或不含药的空白脂质体经乙腈涡旋破乳后离心(
12600 r/min,15 min,4℃),取上清液分别作为供试品溶液和空白溶液;另取TPP-OLA 纯品用流动相稀释作为对照品溶液。 -
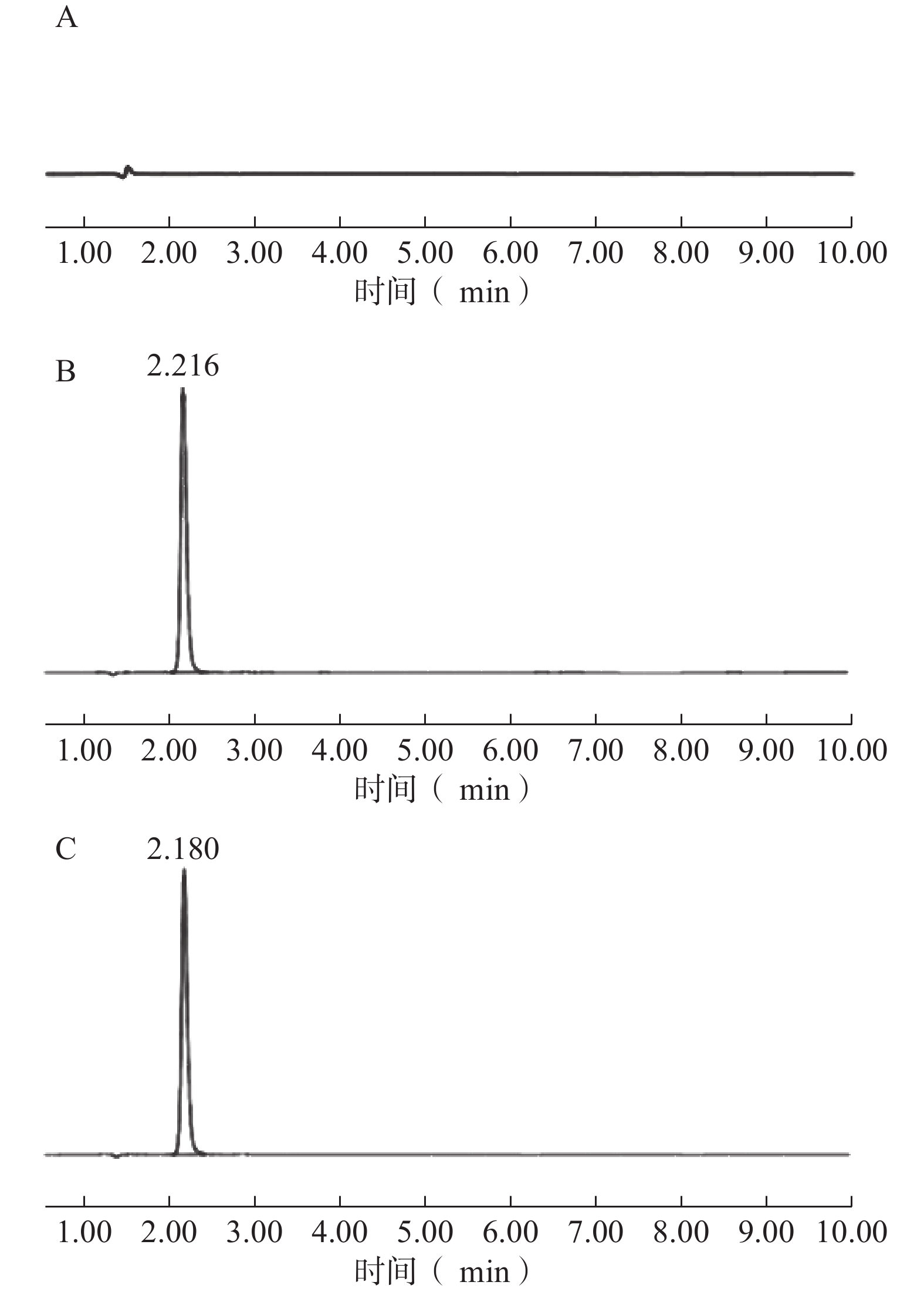
专属性考察表明,在该色谱条件下,空白溶液在TPP-OLA 出峰位置无干扰峰,供试品溶液与对照品溶液的主峰保留时间一致,表明辅料及溶剂对检测无干扰,方法专属性良好(图3)。
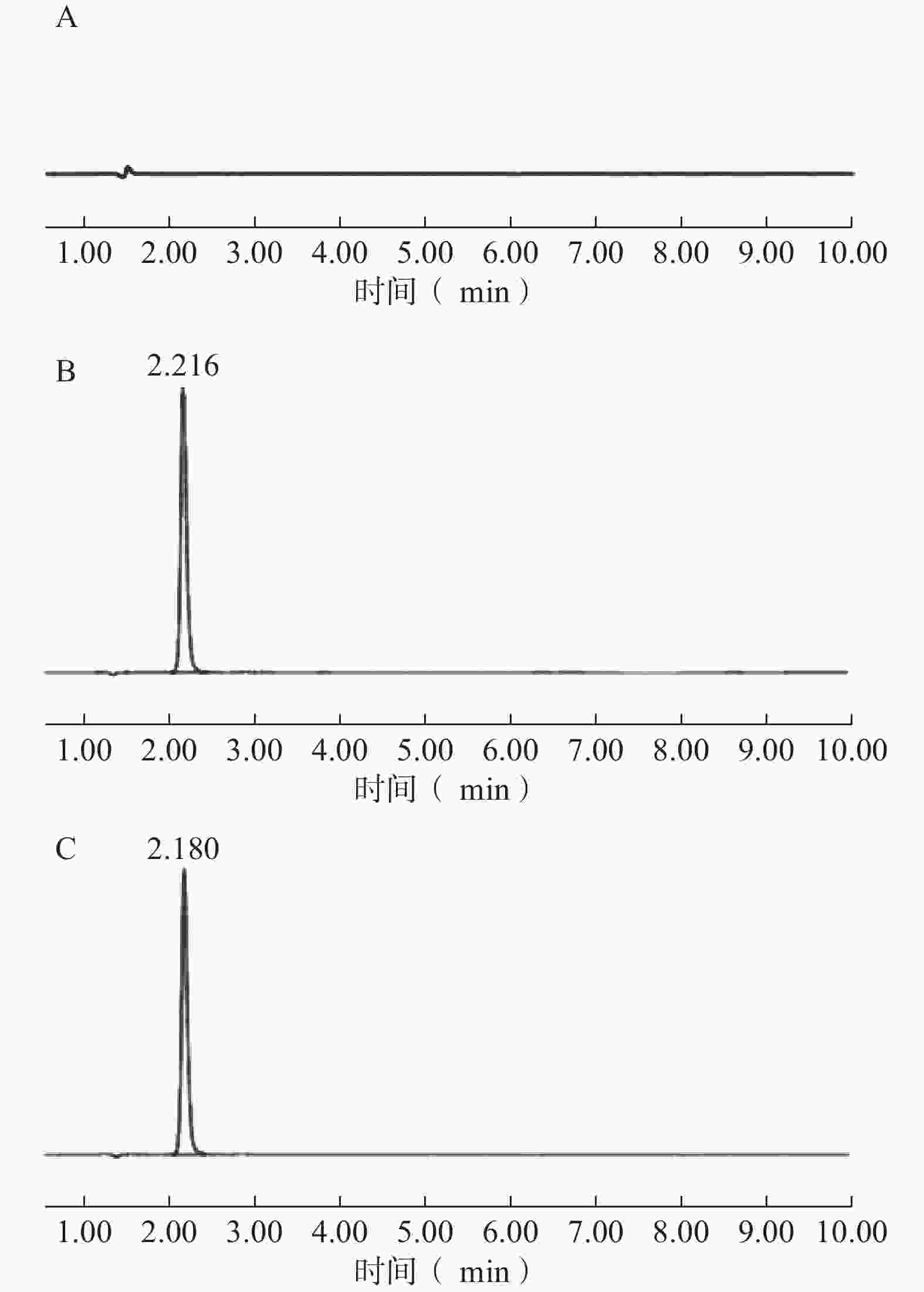
图 3 TPP-OLA的HPLC专属性图谱
-
取TPP-OLA对照品储备液,用流动相逐级稀释,制备浓度在 2~
1024 μg/ml 范围内的系列对照品溶液。按上述色谱条件进样测定,记录吸收峰面积。以TPP-OLA质量浓度(C, μg/ml)为横坐标,相应的吸收峰面积(A, μV·s)为纵坐标进行线性回归。得回归方程为:A = 20203C +44952 (R2 =0.9993 )。结果表明,TPP-OLA 在该浓度范围内线性关系良好。 -
取低、中、高3个浓度(16、128、
1024 μg/ml)的TPP-OLA对照品溶液,分别连续进样分析6次。结果显示,TPP-OLA在上述浓度下的吸收峰面积RSD分别为0.3%、0.2% 和0.2%,符合定量测定要求。 -
取浓度为100 μg/ml的供试品溶液置于室温条件下放置,于24 h内不同时间点进样测定。结果显示,TPP-OLA吸收峰面积的RSD仅为0.1%,表明供试品溶液在室温放置24 h 内化学性质稳定,可满足大批量样本连续自动进样的分析需求。
-
精密量取已知药物含量的TPP-OLA脂质体溶液,分别按本底含量的80%、100%、120%加入TPP-OLA对照品,按照前述条件进行破乳、离心提取上清液后进样测定。结果显示,低、中、高3个浓度水平的平均加样回收率处于98.50%~101.20%之间,RSD分别为2.16%、1.86%和2.30%,表明本研究所建方法具有良好的准确度。
-
将TPP-OLA对照品溶液逐步稀释并进样分析,以信噪比S/N ≈ 3为限度,确定本方法的检测限为0.06 μg/ml;以信噪比S/N ≈ 10为限度,确定本方法的定量限为0.2 μg/ml。该结果表明本方法具有较高的检测灵敏度。
-
本研究固定了脂质骨架的总量及比例,重点对模型药物TPP-OLA的投料量及铺膜溶剂进行了单因素考察。
-
药脂比是决定脂质体包封率和最终载药量的核心参数。本研究分别考察了1、2.5、5、7.5及10 mg药物投料量对脂质体理化性质的影响。结果表明,当投料量处于较低水平(1~5 mg)时,脂质体的包封率均保持在83.3%以上的高水平,且粒径控制在95 nm以下,体系呈现出良好的均一性与稳定性。然而,当投料量进一步增加至7.5 mg和10 mg 时,包封率出现了明显的下降(降至80%以下),表明药物的加入量已逐渐达到该脂质骨架的负载饱和极限。综合考量制剂的绝对载药量与体系稳定性,最终确定5 mg为最佳的TPP-OLA投料量。
-
在确定5 mg最佳投料量的基础上,进一步考察了不同比例的三氯甲烷及三氯甲烷-甲醇混合体系。结果表明,相较于纯三氯甲烷体系,引入甲醇会导致成膜状态不佳,进而使包封率出现明显下降(75.0%~78.0%)。而在纯三氯甲烷体系的对比中,使用6 ml三氯甲烷制备的脂质体,其平均粒径(82.24 nm)明显优于12 ml 组(93.37 nm),且多分散系数(PDI = 0.239)更低,粒径分布更加集中。虽然6 ml组的包封率(80.0%)较12 ml组(83.3%)有轻微回落,但仍属于理想的包封水平。此外,采用较少体积的有机溶剂不仅有利于缩短旋蒸时间、提高制备效率,更能有效降低最终制剂中有机溶剂残留的风险。综合评估,最终确定采用6 ml纯三氯甲烷作为最优铺膜溶剂体系。
-
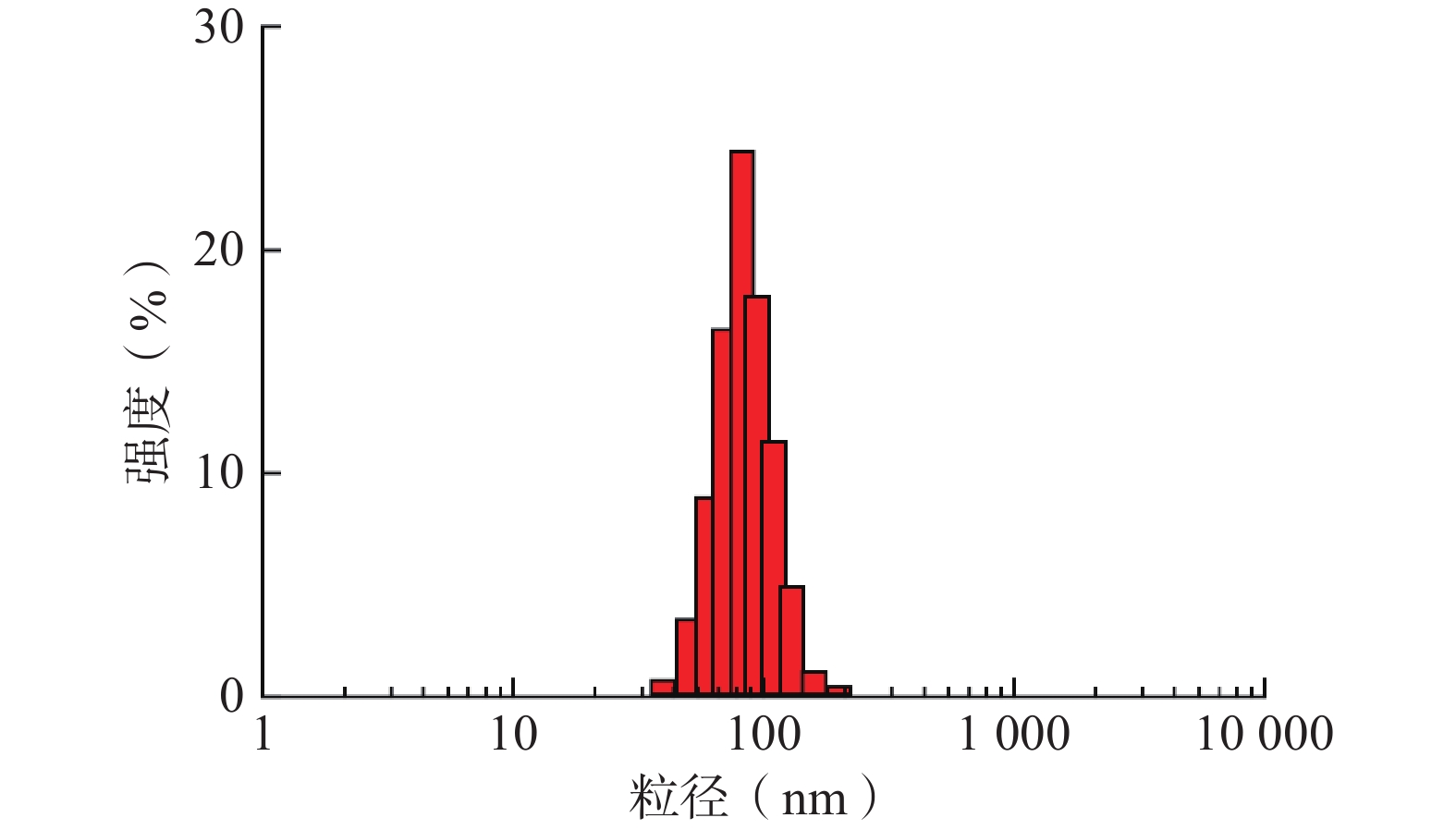
采取上述最优工艺条件制备的TPP-OLA/Lip呈现出良好的单峰分布,其平均粒径为82.24 nm,PDI为0.239,表明胶体体系高度均一(图4)。此外,该最优处方的包封率为80.0%,平均Zeta电位为 + 9.9 mV,表明脂质体表面带有一定的正电荷,可通过粒子间的静电斥力有效维持体系的物理稳定性。
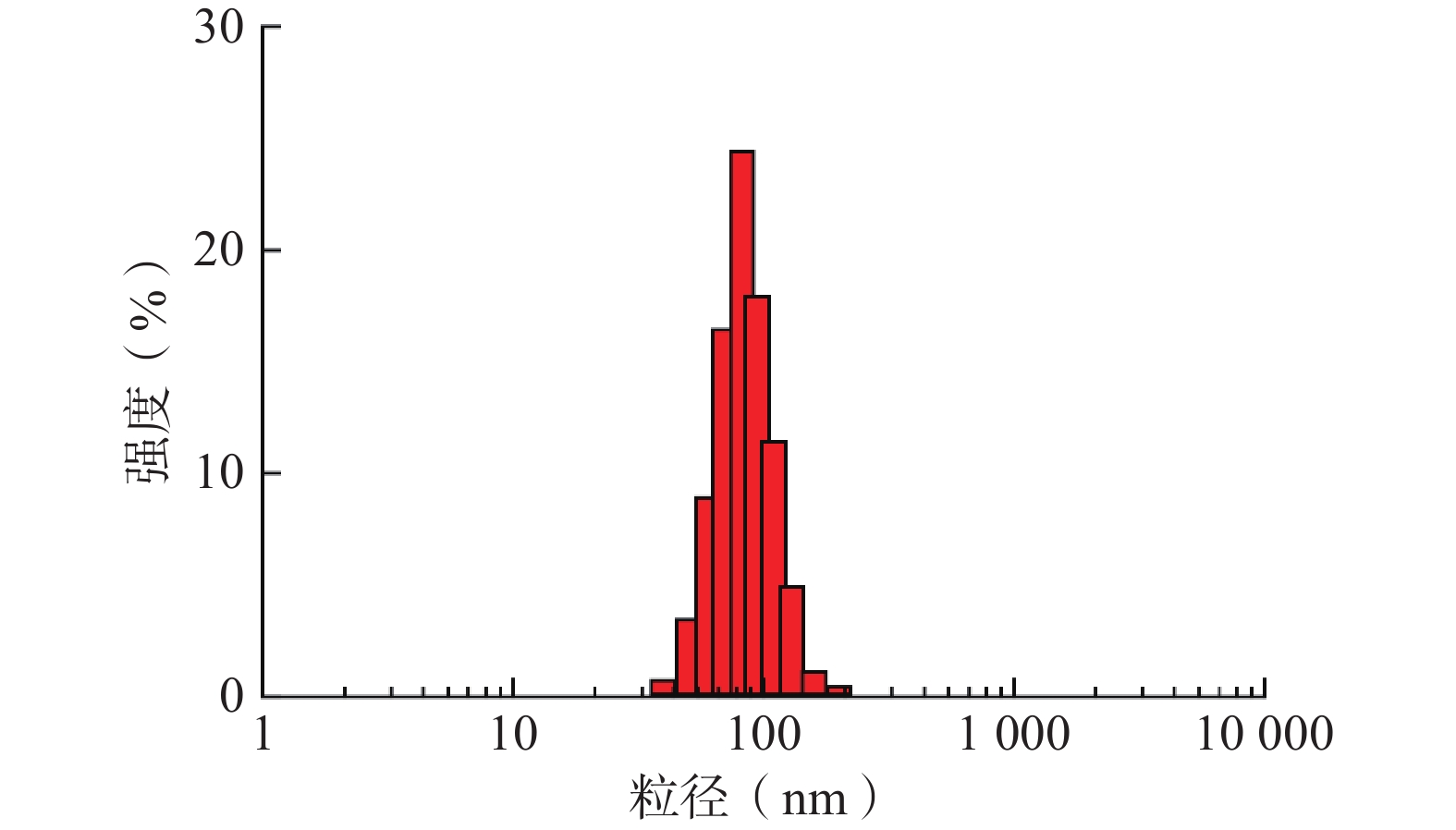
图 4 最优处方TPP-OLA/Lip的粒度分布图
-
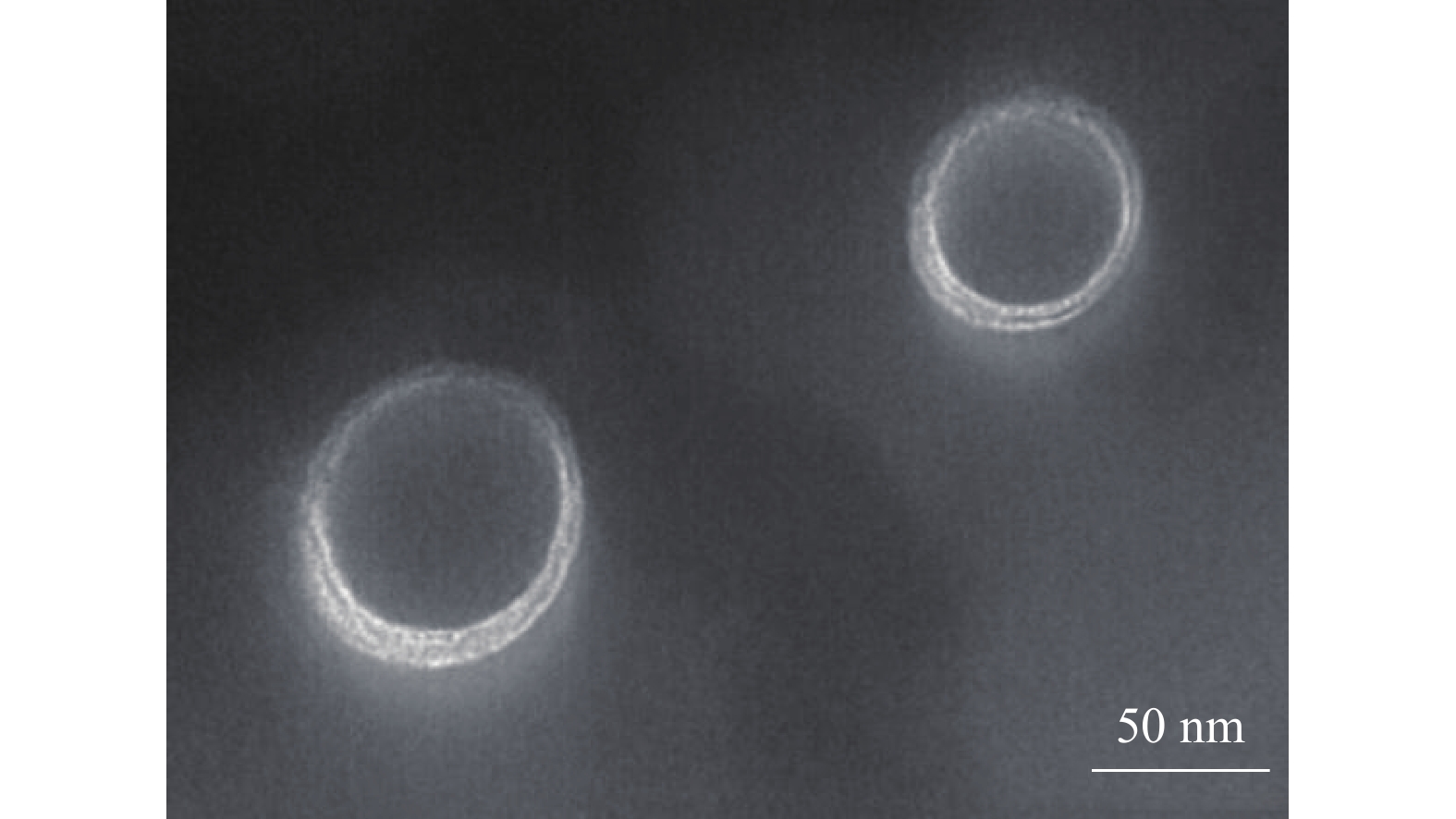
经2%磷钨酸负染后置于透射电镜下观察(图5),TPP-OLA/Lip 呈现出典型的单室脂质体结构,边缘具明显光晕,形态均一、圆整。

图 5 TPP-OLA/Lip的透射电镜图
-
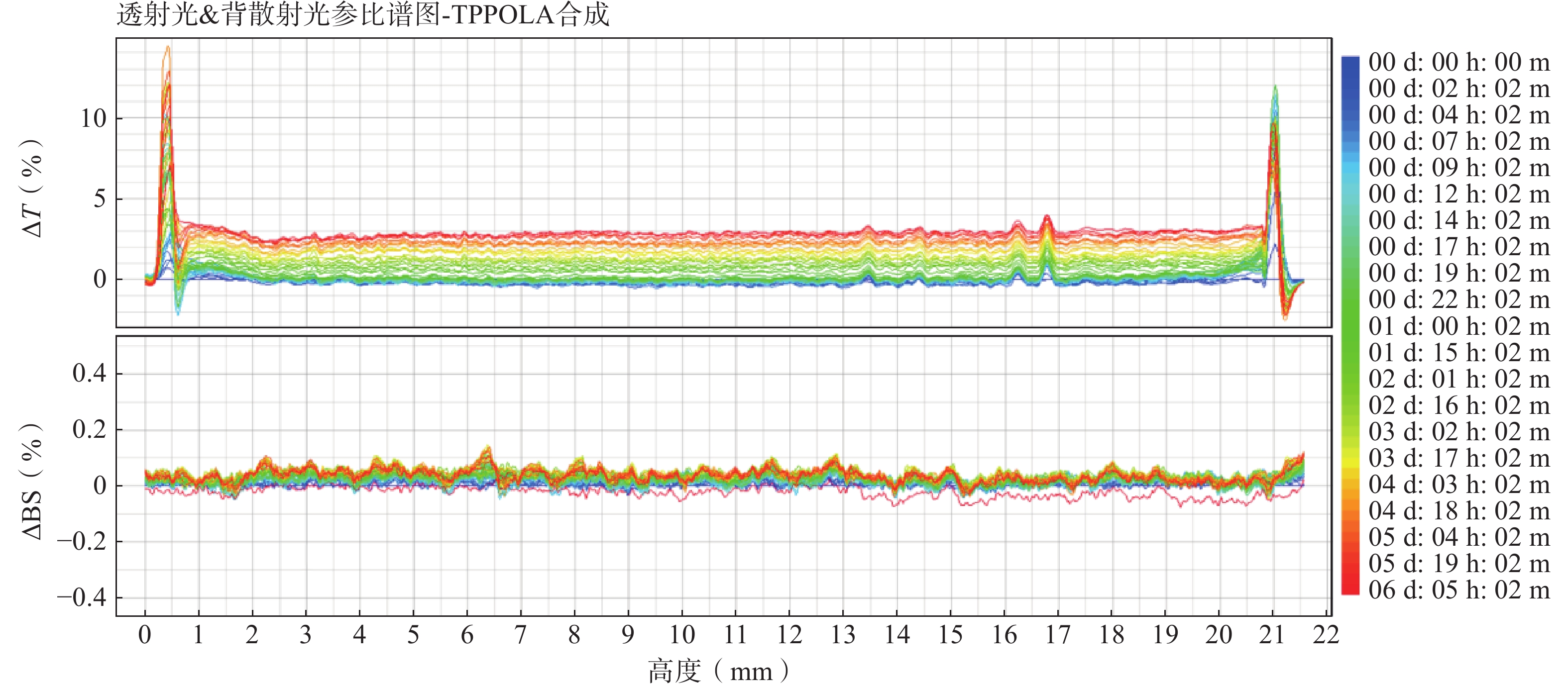
随着连续6天的监测(图6),体系中的ΔT图谱仅发生极其微小且均匀的正向漂移(最大波动幅度小于3%),且ΔBS图谱波动几乎贴近于基线。表明TPP-OLA/Lip体系未发生明显的颗粒聚集或聚沉现象,具有优异的物理稳定性。
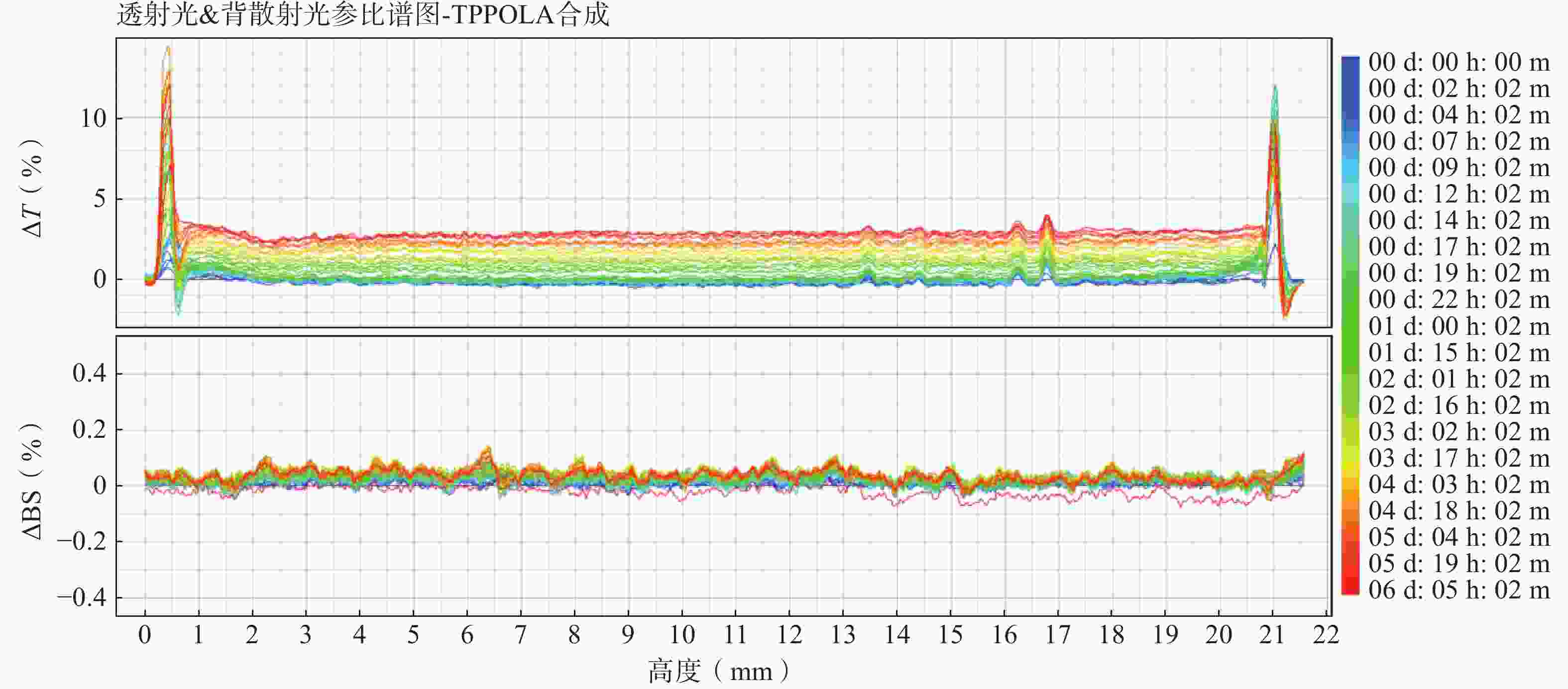
图 6 TPP-OLA/Lip的稳定性分析测定结果
-
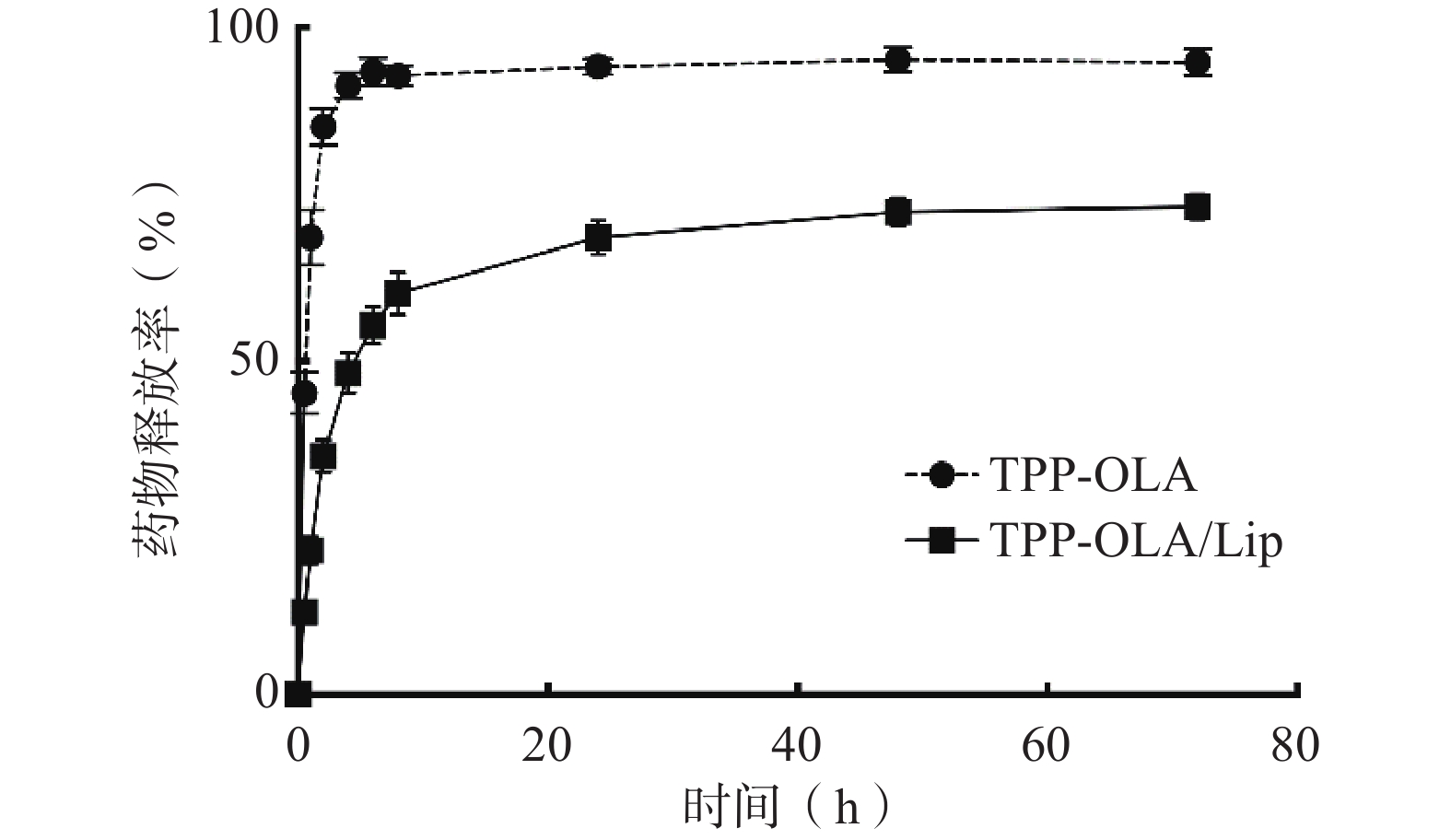
游离TPP-OLA分子在4 h内累积释放率达到91.3%,表现出明显的突释现象(图7)。而TPP-OLA/Lip在0~4 h 内有约48.2%的初始释放,随后呈现平稳缓慢释药,72 h最终累积释药率为73.1%。
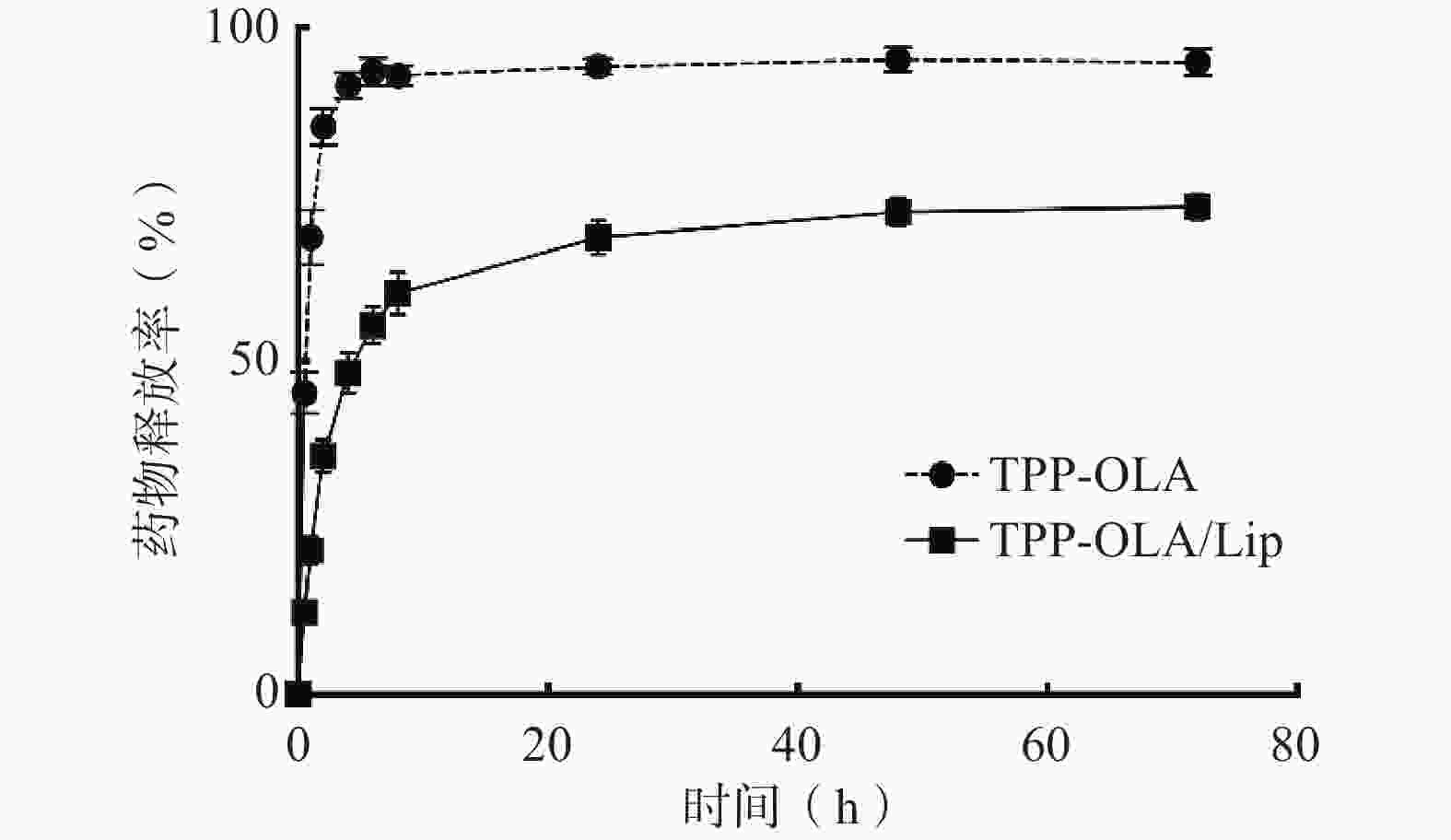
图 7 TPP-OLA和TPP-OLA/Lip的体外累积释放曲线
-
TBI继发性损伤中的能量耗竭与线粒体PARP的过度激活密切相关。然而,传统PARP抑制剂(如奥拉帕尼)受限于水溶性差及缺乏亚细胞靶向性,难以在损伤部位的线粒体内达到有效治疗浓度。针对此困境,本研究设计并合成了一种新型线粒体靶向抑制剂TPP-OLA,并成功构建了其纳米脂质体递送系统,对其理化性质进行了系统评价。
研究结果表明,TPP-OLA成功实现了纳摩尔级别的PARP酶高效抑制(IC50 = 4.1 nM)。同时,实验数据确证,经处方工艺优化的TPP-OLA脂质体形态圆整、粒径分布均匀(约 82.24 nm),包封率达80.0%,且表面带有适宜的正电荷(+9.9 mV),赋予了体系优良的物理稳定性。
本研究的核心优势在于提出了“化学修饰 + 纳米递送” 的联合给药策略。在分子层面,通过亲脂性阳离子三苯基膦(TPP)的共价修饰,赋予了原药精准的线粒体靶向性。该设计有望在精准抑制异常线粒体PARP激活、阻断能量耗竭进程的同时,避免干扰细胞核内正常的生理修复功能,为靶向神经保护治疗提供了极具前景的分子设计思路。
综上所述,本研究通过合理的结构修饰与剂型优化,成功开发出具有优良成药潜力的TPP-OLA及其脂质体递送系统。未来研究可进一步考察该系统在TBI动物模型中的跨血脑屏障递送效率、体内药代动力学及抗凋亡药效,为新型神经保护药物的开发奠定基础。鉴于TPP-OLA优异的理化性质与包载适应性,后续亦可尝试将其载于聚乳酸-羟基乙酸共聚物(PLGA)等高分子聚合物载体中,以探索不同微环境下的释药动力学与长效干预策略。
Synthesis of mitochondria-targeted Olaparib and preparation of brain-targeting liposomes
-
摘要:
目的 合成线粒体靶向聚ADP-核糖聚合酶(PARP)抑制剂,并制备其脑靶向脂质体以考察理化性质,为创伤性脑损伤(TBI)治疗提供新策略。 方法 将奥拉帕尼与三苯基膦(TPP)偶联制备TPP-OLA,经波谱学确证结构并评价其体外酶学抑制活性。建立高效液相色谱法(HPLC)用于定量分析,采用薄膜水化法制备脑靶向载药脂质体并优化工艺,考察其体外释药行为。 结果 成功获得高纯度TPP-OLA,其对PARP酶的半数抑制浓度为4.1 nM,保留了相应的酶抑制活性。建立的HPLC法在2~ 1024 μg/ml浓度范围内线性良好(R2=0.9993 )。优选处方制备的脑靶向脂质体外观圆整,平均粒径82.24 nm,平均Zeta电位为 + 9.9 mV,包封率达80.0%,体外物理稳定性良好,且展现出长达72 h的平稳缓释特征。结论 TPP-OLA合成路线可行,其载药脂质体具备适宜的理化性质与成药性,为TBI的神经保护治疗提供了递送载体。 Abstract:Objective To synthesize a mitochondria-targeted poly (ADP-ribose) polymerase (PARP) inhibitor and prepare its brain-targeted liposomes to investigate their physicochemical properties, aiming to provide a novel strategy for the treatment of traumatic brain injury (TBI). Methods Olaparib was conjugated with triphenylphosphonium (TPP) to synthesize TPP-OLA. The chemical structure was confirmed by spectroscopic analysis, and its enzymatic inhibitory activity was evaluated in vitro. A high-performance liquid chromatography (HPLC) method was established for quantitative analysis. Brain-targeted drug-loaded liposomes were prepared using the thin-film hydration method. The preparation process was optimized, and their in vitro release behavior was investigated. Results High-purity TPP-OLA was successfully obtained, exhibiting a half-maximal inhibitory concentration (IC50) of 4.1 nM against the PARP enzyme, thereby retaining the corresponding enzyme inhibitory activity. The established HPLC method demonstrated good linearity within the concentration range of 2– 1024 μg/ml (R2=0.9993 ). The brain-targeted liposomes prepared using the optimized formulation displayed a well-rounded morphology, an average particle size of 82.24 nm, an average Zeta potential of +9.9 mV, an encapsulation efficiency reaching 80.0%, favorable in vitro physical stability, along with a steady sustained-release profile over 72 h.Conclusion The synthetic route for TPP-OLA is feasible. The resulting drug-loaded liposomes possess suitable physicochemical properties and druggability, offering a novel delivery vehicle for precise neuroprotective therapy of TBI. -
Key words:
- traumatic brain injury /
- mitochondrial targeting /
- Olaparib /
- PARP inhibitor /
- liposomes
-
[1] ZHANG L, WANG H D, ZHOU X M, et al. Role of mitochondrial calcium uniporter-mediated Ca2+ and iron accumulation in traumatic brain injury[J]. J Cellular Molecular Medi, 2019, 23(4): 2995-3009. doi: 10.1111/jcmm.14206 [2] MODI H R, MUSYAJU S, RATCLIFFE M, et al. Mitochondria-targeted antioxidant therapeutics for traumatic brain injury[J]. Antioxidants, 2024, 13(3): 303. doi: 10.3390/antiox13030303 [3] JIANG J Y, GAO G Y, FENG J F, et al. Traumatic brain injury in China[J]. Lancet Neurol, 2019, 18(3): 286-295. doi: 10.1016/S1474-4422(18)30469-1 [4] 韩亚进, 孙科, 潘卫民, 等. 1990—2023年中国创伤性颅脑损伤疾病负担与变化趋势分析[J]. 中国修复重建外科杂志, 2025, 39(11): 1388-1394. [5] HACKENBERG K, UNTERBERG A. Schädel-Hirn-Trauma: Traumatic brain injury[J]. Nervenarzt, 2016, 87(2): 203-216. [6] YONUTAS H M, VEKARIA H J, SULLIVAN P G. Mitochondrial specific therapeutic targets following brain injury[J]. Brain Res, 2016, 1640(Pt A): 77-93. [7] SATCHELL M A, ZHANG X P, KOCHANEK P M, et al. A dual role for poly-ADP-ribosylation in spatial memory acquisition after traumatic brain injury in mice involving NAD+ depletion and ribosylation of 14-3-3γ[J]. J Neurochem, 2003, 85(3): 697-708. doi: 10.1046/j.1471-4159.2003.01707.x [8] SZCZESNY B, BRUNYANSZKI A, OLAH G, et al. Opposing roles of mitochondrial and nuclear PARP1 in the regulation of mitochondrial and nuclear DNA integrity: implications for the regulation of mitochondrial function[J]. Nucleic Acids Res, 2014, 42(21): 13161-13173. doi: 10.1093/nar/gku1089 [9] 皮如玉, 李佳蕊. PARP抑制剂在卵巢癌治疗中的研究进展[J]. 国际妇产科学杂志, 2016, 43(5): 515-518. doi: 10.3969/j.issn.2095-1264.2025.01.03 [10] 王莹颖, 刘文景, 宁瑶, 等. PARP抑制剂的作用机制和研究进展[J]. 中国新药杂志, 2018, 27(3): 306-313. doi: 10.3969/j.issn.1003-3734.2018.03.011 [11] 徐鹏程, 于鑫, 郭兆明, 等. 靶向线粒体的载体给药系统研究进展[J]. 内蒙古大学学报(自然科学版), 2017, 48(2): 219-224. doi: 10.13484/j.nmgdxxbzk.20170219 [12] SUKHANOVA E I, TRENDELEVA T A, ZVYAGILSKAYA R A. Interaction of yeast mitochondria with fatty acids and mitochondria-targeted lipophilic cations[J]. Biochemistry, 2010, 75(2): 139-144. doi: 10.1134/s0006297910020033 [13] MURPHY M P, SMITH R A J. Targeting antioxidants to mitochondria by conjugation to lipophilic cations[J]. Annu Rev Pharmacol Toxicol, 2007, 47: 629-656. doi: 10.1146/annurev.pharmtox.47.120505.105110 [14] LIU Y, HUANG R Q, HAN L, et al. Brain-targeting gene delivery and cellular internalization mechanisms for modified rabies virus glycoprotein RVG29 nanoparticles[J]. Biomaterials, 2009, 30(25): 4195-4202. doi: 10.1016/j.biomaterials.2009.02.051 [15] KRAINZ T, LAMADE A M, DU L N, et al. Synthesis and evaluation of a mitochondria-targeting poly(ADP-ribose)polymerase-1 inhibitor[J]. ACS Chem Biol, 2018, 13(10): 2868-2879. doi: 10.1021/acschembio.8b00423 -

 点击查看大图
点击查看大图
图(7)
计量
- 文章访问数: 322
- HTML全文浏览量: 135
- PDF下载量: 2
- 被引次数: 0
