

 下载:
下载:

-
病毒是一种非细胞的微生物,具有完整的基因组、多种蛋白质和核糖核酸(RNA),表现出强烈的传染性和广泛的流行性。目前,超过80%的呼吸道传染性疾病由病毒引起,并在全球范围内造成了严重的经济和公共卫生挑战[1]。病毒性疾病对人类社会的影响尤为深远。2003年,SARS冠状病毒(SARS-CoV)引起的严重急性呼吸系统综合征(SARS)感染了数千万人[2];2019年,新型冠状病毒(SARS-CoV-2)爆发,导致全球约7.59亿人感染,680万人死亡,疫情蔓延迅速,波及范围广泛,成为百年来最严重的公共卫生危机之一[3],严重扰乱了全球的经济秩序、社会生活和医疗体系,引发了全球范围内的广泛关注[4]。
冠状病毒(CoV)是一类单链正链RNA病毒,根据基因组和抗原特性的差异,可进一步分为4个属:α、β、γ和δ[5]。SARS-CoV-2属于β属冠状病毒,与SARS-CoV和中东呼吸综合征冠状病毒(MERS-CoV)等具有较高的同源性[6]。在SARS-CoV-2的生命周期中,主蛋白酶(Mpro)扮演着至关重要的角色。Mpro又称为3C样蛋白酶(3CLpro),是一种半胱氨酸蛋白酶,负责切割病毒编码的多聚蛋白,产生具有生物活性的病毒蛋白,在病毒的复制和转录过程中起着至关重要的作用[7]。由于Mpro在不同冠状病毒中高度保守,且在人类细胞中没有同源蛋白,因此成为抗病毒药物研发的理想靶点[8]。本文综述了近年来以Mpro为靶点的抑制剂,包括拟肽类共价抑制剂、非拟肽类共价抑制剂和非共价类抑制剂的研究进展,还探讨了靶向Mpro 的蛋白降解靶向嵌合体(PROTAC)降解剂的创新研究策略,为未来抗病毒药物的研发提供新思路和新方向。
-
由于Mpro在SARS-CoV-2的复制转录过程中的关键作用及其在不同冠状病毒中的高度保守性,研发靶向Mpro的抑制剂成为新冠病毒治疗的重要方向。根据化学结构和作用机制的不同,Mpro抑制剂主要分为3类:拟肽类共价抑制剂、非拟肽类共价抑制剂和非共价类抑制剂。
-
通过对病毒多聚蛋白切割位点的序列分析及突变实验发现,Mpro的切割位点位于Gln(谷氨酰胺)处。晶体结构研究表明,Mpro的S1亚位点能够精确容纳Gln的侧链结构,而其他氨基酸因存在空间位阻或缺乏互补作用,难以与Mpro结合。此外,天然多肽底物在体内易被蛋白酶降解,稳定性较差,因此,需通过化学修饰提高其稳定性[9]。基于此设计拟肽类共价抑制剂的策略是保留Gln的结合位点,并对周边序列进行优化,形成特异性共价结合基团(弹头)实现对酶活性的高效抑制。目前这类抑制剂包括α-酮酰胺类、肽醛类、羟甲基酮类、噻唑烷酮类和氰基类拟肽化合物等[10]。
-
Zhang等[11-12]基于谷氨酰胺的衍生物设计合成含有α-酮酰胺结构的化合物1(图1)。该化合物通过α-酮基亲核进攻Mpro活性位点的Cys145,形成硫代半缩酮结构,从而发挥酶活抑制作用。因此,这类化合物被称为α-酮酰胺类拟肽抑制剂。α-酮酰胺类抑制剂与Mpro活性位点半胱氨酸残基形成的是一种可逆共价键,其中α-酮基氧和酰胺基氧可作为氢键受体,与Mpro中的氨基酸残基Gly143和Cys145形成氢键相互作用,从而进一步增强抑制剂与酶的结合能力。化合物1对多种病毒的Mpro表现出显著的抑制活性,对SARS-CoV、HCoV-NL63、CVB3这3种病毒Mpro的IC50分别为1.95、6.6和1.2 μmol/L,抗病毒活性EC50分别为4.5、4.5和2.0 μmol/L。因此,化合物1是一种α型、β型冠状病毒和肠道病毒Mpro的广谱抑制剂。为了提高化合物1在血浆中的暴露量,研究人员对其结构进行优化,将P3部分疏水性肉桂酰基替换为Boc基团,并将P2部分苯环替换为环丙基甲基,得到化合物2 (IC50=0.67 μmol/L, EC50=4.0 μmol/L)。药代动力学研究表明,以20 mg/kg剂量对小鼠进行皮下给药时,化合物2在血浆中停留时间较短,仅4 h,血浆半衰期为1.8 h。而在给药24 h后,肺组织中的药物含量仍维持在约135 ng/g。由于冠状病毒主要影响肺组织,该化合物在肺组织中呈现出长时间的高浓度滞留特性,其分布特征可能对冠状病毒感染的治疗具有积极意义。
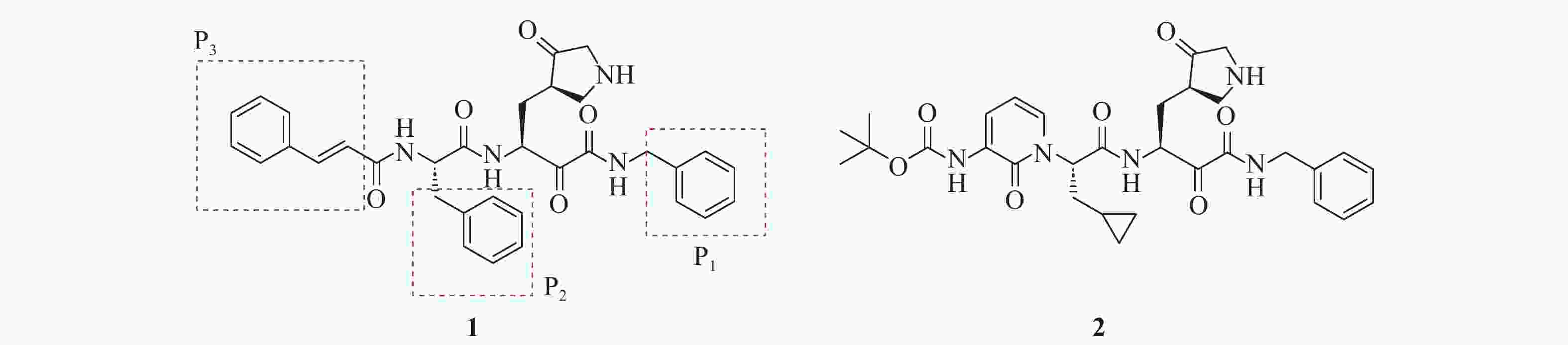
图 1 α-酮酰胺类抑制剂
-
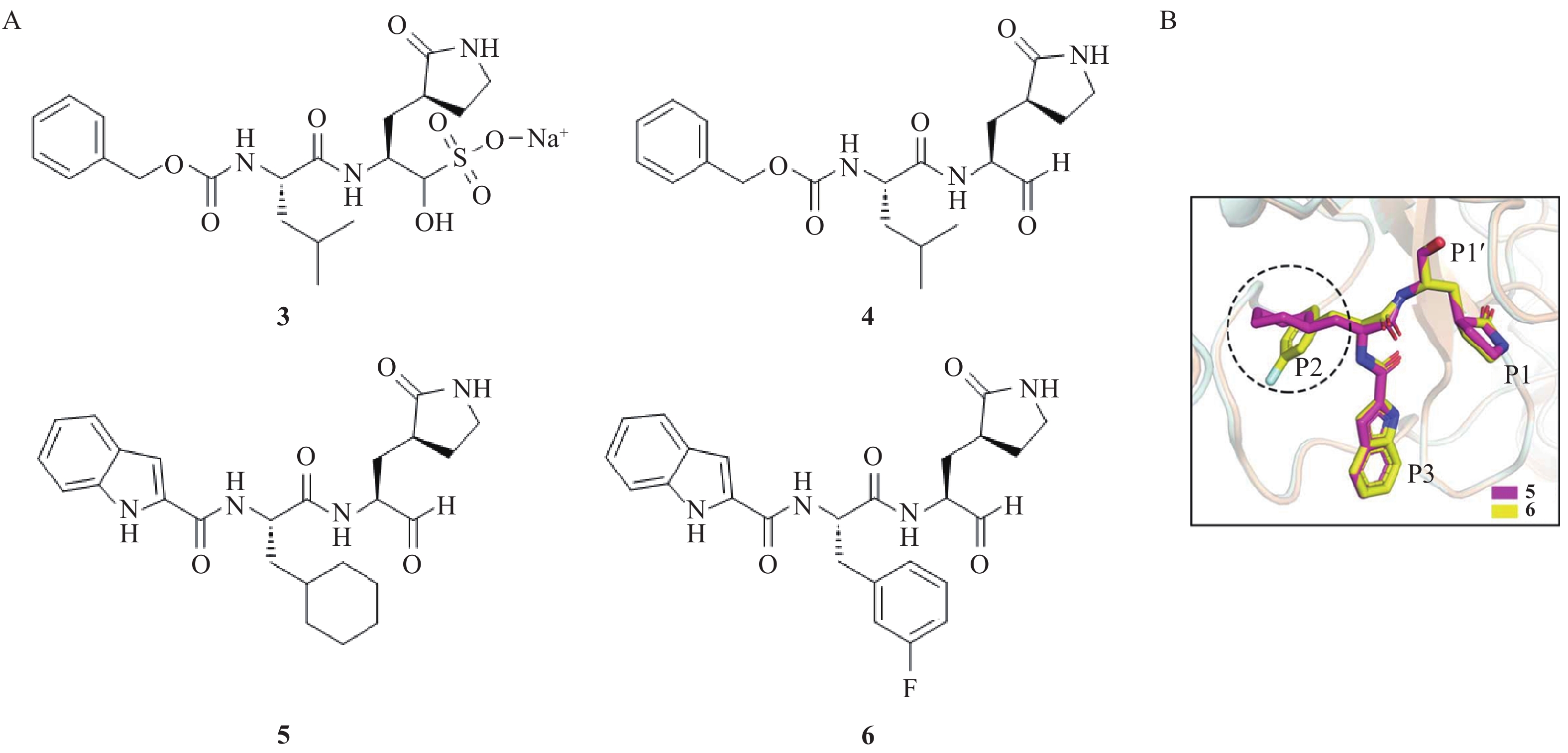
将α-酮酰胺类抑制剂中的α-酮酰胺替换为醛基,可得到肽醛类抑制剂。醛基与Mpro的Cys145的巯基形成不可逆的硫代半缩醛共价键,从而增强了抑制剂与靶蛋白的结合亲和力[13]。基于这一策略,Ma等[14]设计并合成了化合物3和4(图2),二者表现出较强的Mpro抑制活性,IC50值分别为30 nmol/L和400 nmol/L,且对宿主蛋白酶无明显抑制作用,显示出极高的选择性。化合物3的醛基以双硫酸盐形式存在,通过可逆加成的方式保护醛基,显著提高了化合物的水溶性和稳定性,更加便于体内给药[15-16]。在体内,化合物3经酶催化后转化为化合物4,其暴露的α-醛基能够与Mpro活性位点的Cys145形成不可逆共价结合。Dai等[17]综合生物利用度和毒性,以化合物3为先导化合物设计并合成一系列带有醛基共价弹头的化合物。其中,化合物5和6的(S)-γ-内酰胺环占据Mpro的深疏水S2口袋,并与周围氨基酸His41和Gln189等残基形成氢键相互作用,环己基和3-氟苯基可以深入蛋白酶空腔,并于周围的Met165、Met49、Val186以及Arg188残基形成疏水相互作用。药代动力学研究显示,化合物5经小鼠腹腔和静脉给药后,半衰期分别为5.21 h和1.65 h,腹腔给药的生物利用度达到80%以上。化合物6以5.0 mg/kg剂量分别通过小鼠腹腔和静脉给药,半衰期分别为4.27 h和4.41 h,其中腹腔给药时生物利用度达到87.8%,清除率为17.4 ml/(min·kg)。该化合物目前正处于临床试验阶段,显示出良好的开发前景。
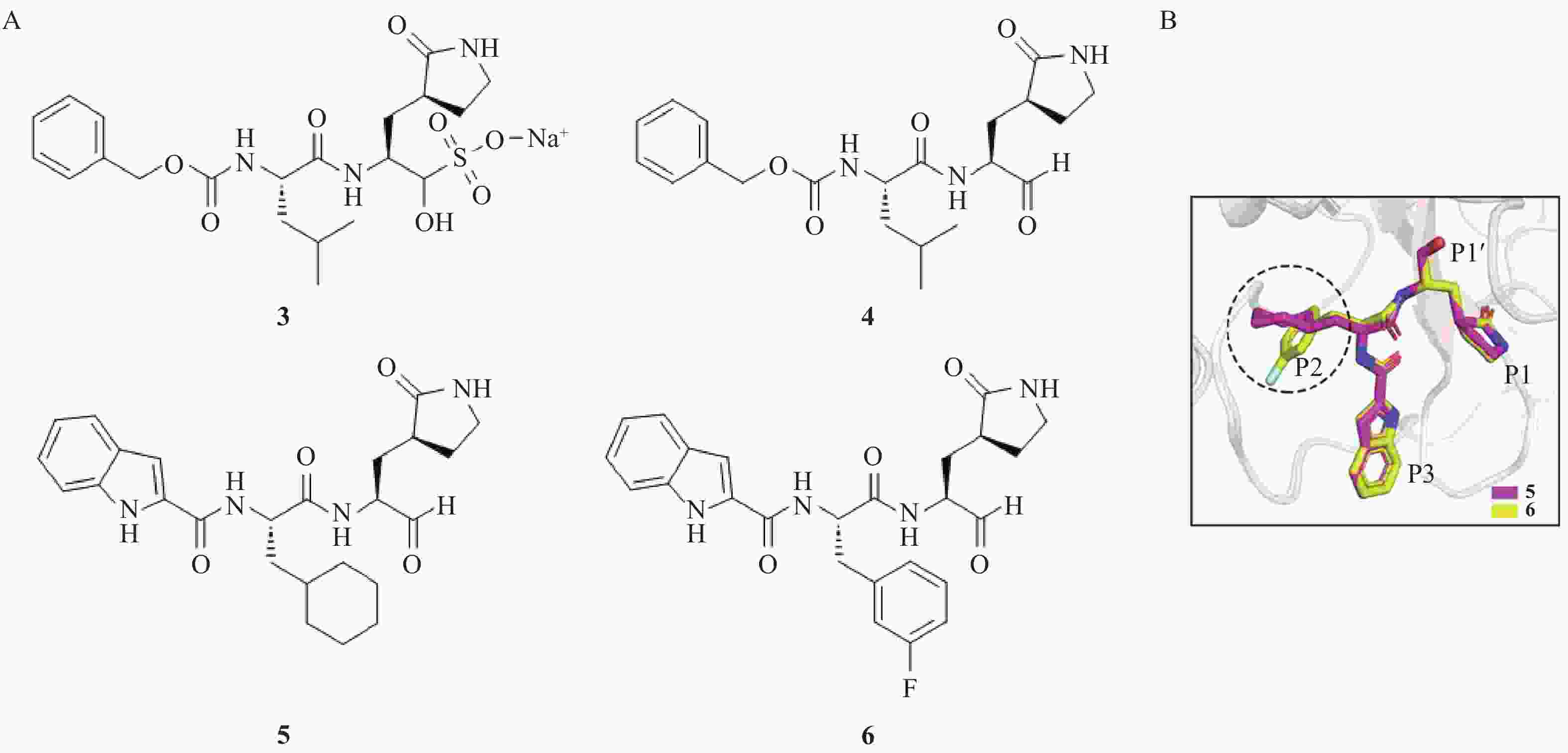
图 2 肽醛类抑制剂
-
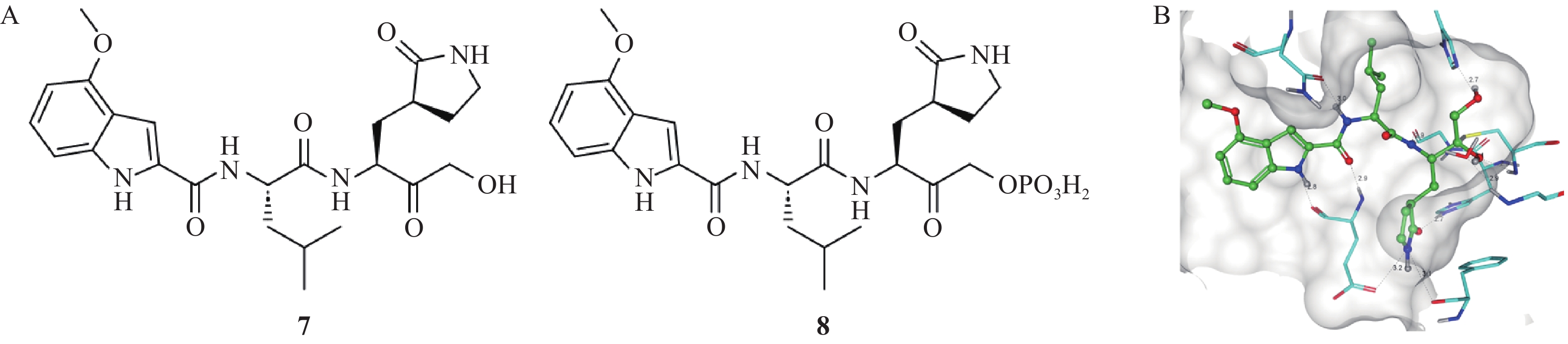
由辉瑞公司设计合成的PF-00835231(化合物7,图3)是一种有效的SARS-CoV-2 Mpro抑制剂[18]。它是基于羟甲基酮弹头设计的Mpro共价抑制剂,该化合物作用机制是羟甲基酮弹头的羰基碳与Mpro活性位点Cys145上的巯基发生反应,进而生成四面体甲醇复合物[19];同时甲醇羟基还通过一个桥连水分子与Cys145主链氮和Gly143酰胺氮形成氢键相互作用网,进一步稳固抑制剂-酶复合物,这种协同作用使得抑制剂的酶活抑制作用显著增强。化合物7能够在纳摩尔级浓度下有效抑制Mpro的活性,抑制常数Ki为0.27 nmol/L,IC50为4.0 nmol/L。体外抗病毒实验显示,化合物7在Vero76细胞模型中的EC50值为0.11 μmol/L。大鼠药代动力学研究显示,静脉给药后,化合物7的血浆清除率为28.6 ml/(min·kg),消除半衰期为1.0 h。SARS-CoV-MA15感染的BALB/c小鼠模型实验结果显示,在30~300 mg/kg剂量范围内,化合物7呈剂量依赖性地降低肺组织内病毒滴度,肺部组织学病理变化证实药物能够减轻肺部病变。为进一步优化药物性能并改善其体内药物清除率,研究人员设计并合成了磷酸酯前药化合物8(IC50=6.8 nmol/L),显著提高药物的水溶性和生物利用度,为临床应用提供更多便利[20]。
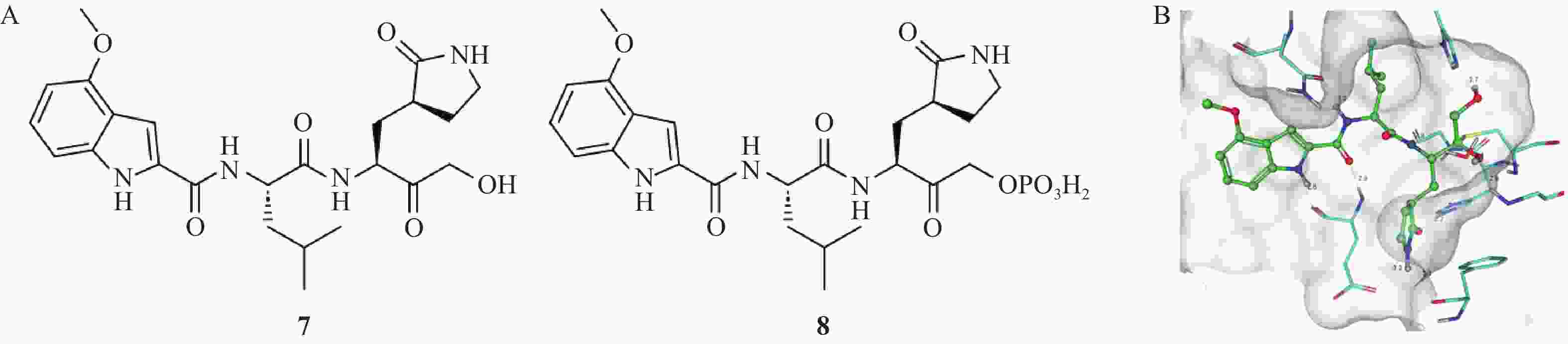
图 3 羟甲基酮类抑制剂
-
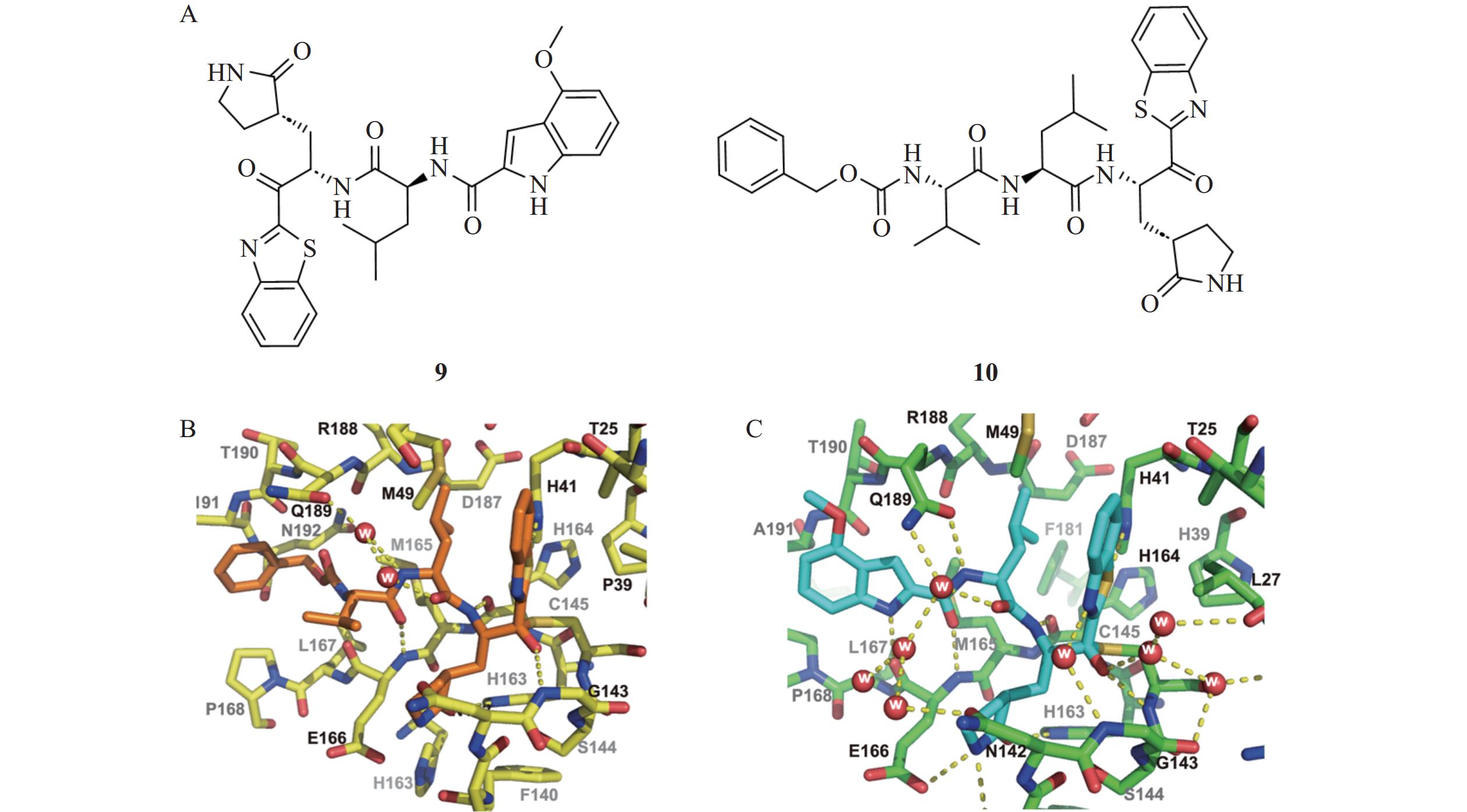
Konno等[10]设计开发了一类以苯并噻唑酮为共价弹头的新型Mpro抑制剂(图4)。这类化合物通过亲电酮基与Mpro的活性位点Cys145发生可逆共价结合,形成四配位半硫酮键,从而抑制Mpro的催化活性。其中,化合物9(Ki=34.7 nmol/L)和化合物10(Ki=14.5 nmol/L)展现出优秀的Mpro抑制活性。化合物9与Mpro的复合物共晶结构解析表明,化合物主链与Mpro的第十二条β链呈反向平行结合模式,苯并噻唑的氮原子和硫原子通过水分子介导与His41形成氢键相互作用,显著增强了化合物的结合活性。此外,化合物9的甲氧基吲哚基团能够诱导Mpro发生构象重排,一方面导致酶活性位点处空间收缩,另一方面引起环区位移,从而使活性位点呈现更加闭合的状态。这种独特的构象变化进一步增强了化合物9与Mpro的结合能力,而在目前已报道的Mpro抑制剂中尚未观察到类似现象。因此,化合物9作为一种较小的二肽型抑制剂,在不增加氮末端基团的情况下,通过改变环区构象实现了更紧密的结合,并与Mpro保持较强的相互作用。这一特性使其在抑制剂设计中具有重要的创新意义和参考价值,为其进一步优化和开发提供了新的思路和方向。
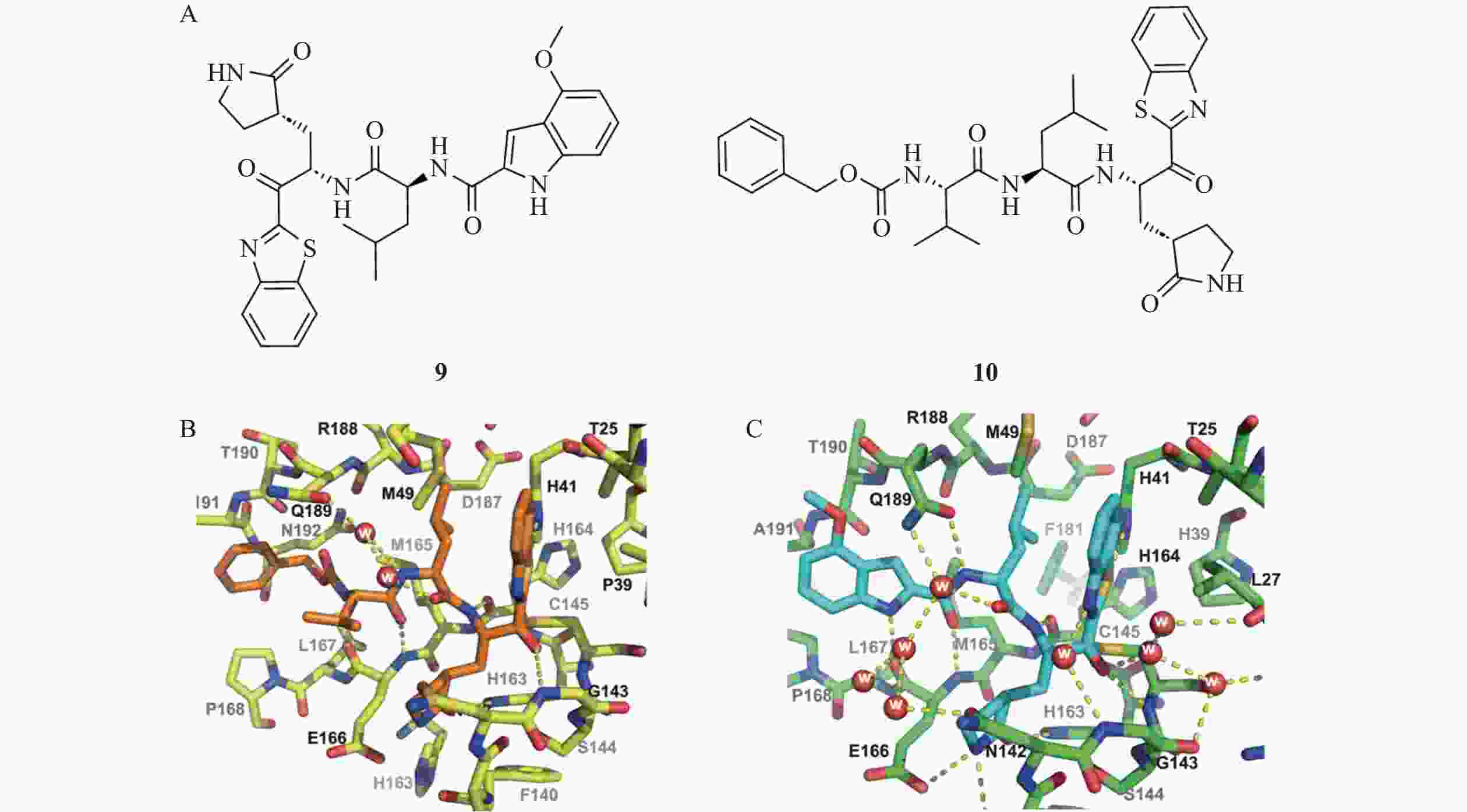
图 4 噻唑烷酮类抑制剂
-
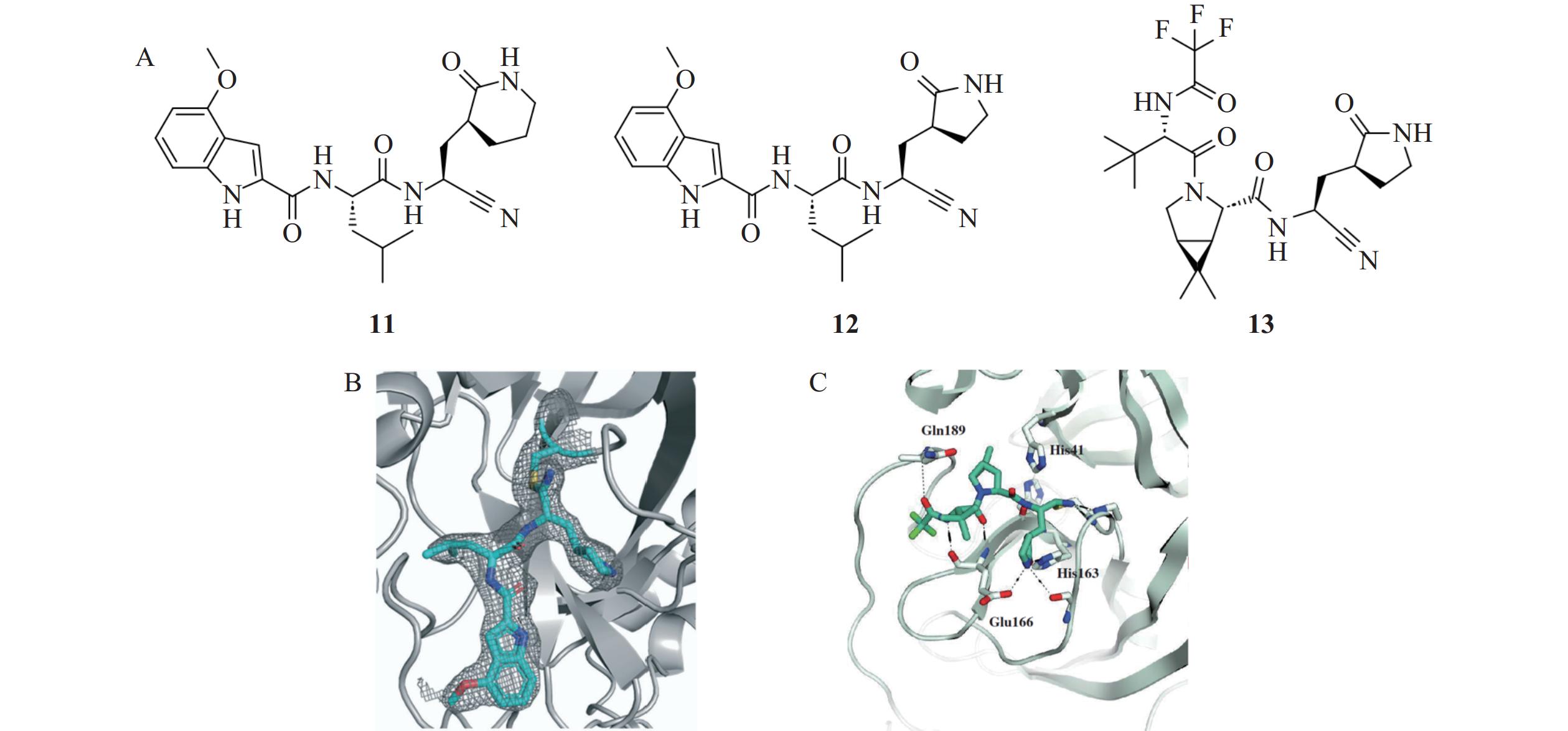
氰基是一种亲电性的化学基团,能与Mpro活性位点关键残基Cys145中的巯基发生共价结合,形成硫代亚胺酯加成物[21]。基于这一机制,Bai等[22]靶向Mpro活性位点的关键残基Cys145,设计并合成了含有氰基弹头的新型共价抑制剂11和12(图5)。通过基于荧光共振能量转移(FRET)的酶抑制实验发现,氰基类共价抑制剂对Mpro表现出良好的抑制效果,且选择性显著优于肽醛类抑制剂。构效关系研究发现,含有δ-内酰胺的化合物11(IC50=13 nmol/L)对Mpro的抑制活性优于含有γ-内酰胺的化合物12(IC50=40 nmol/L)。此外,化合物结构中的吲哚环取代基改变对生物活性影响较大,例如当取代基为4-甲氧基时,目标化合物的疏水性增强,并与Mpro的结合活性也显著提升。然而,在SARS-CoV-2噬斑减少实验中发现,该类化合物的抗病毒活性受到细胞外排泵的影响。加入外排泵抑制剂CP-100356后,化合物的抗病毒活性得到显著提高,表明外排机制是影响其抗病毒活性的关键因素,这一发现为进一步优化该类化合物的药代动力学性质提供了重要线索。
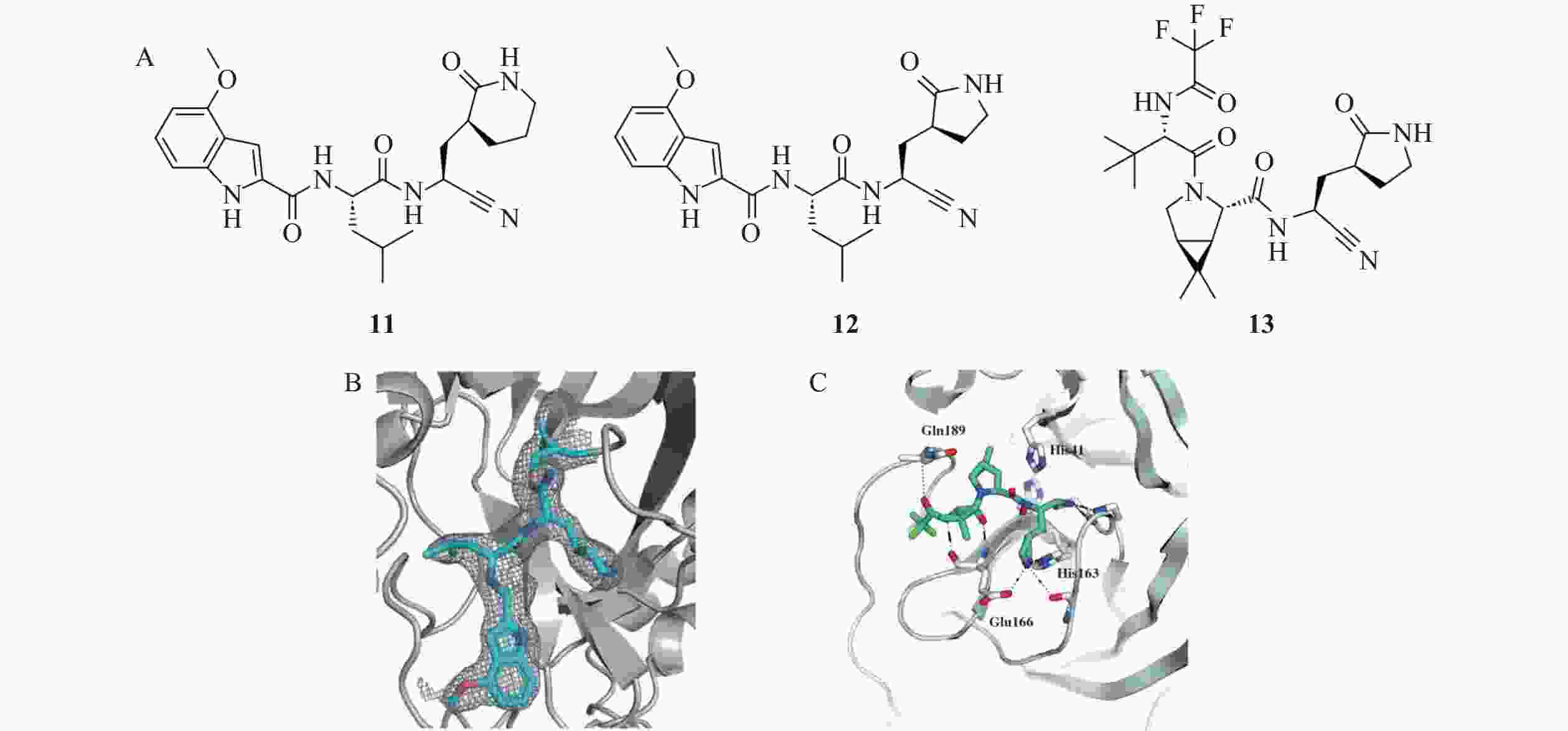
图 5 氰基类抑制剂
研究发现氰基不仅在提高Mpro抑制活性方面具有优势,还可用于提高抗病毒药物的口服生物利用度。Owen等[23]对化合物7进行了结构优化,用氰基替换原有的羟甲基酮结构,在增强与Mpro共价结合的同时,有效降低化合物与血浆蛋白的相互作用,显著提升口服生物利用度。随后,研究人员将吲哚基替换为三氟乙酰酰胺后,得到化合物13(奈玛特韦),其在肝微粒体的代谢稳定性显著提高,固有清除率明显下降。化合物13对Mpro表现出纳摩尔级的抑制活性(IC50=18 nmol/L),并在体外抗病毒实验中展现出优异的活性(EC50=74.5 nmol/L)。此外,通过MDCK-LE细胞渗透性实验发现,化合物13的渗透率为1.1×10−6 cm/s,表明三氟乙酰酰胺基团的引入有效增强了化合物的细胞膜穿透能力,这对于口服吸收和提升生物利用度至关重要。化合物13目前已获得FDA批准用于SARS-CoV-2感染治疗。Ⅲ期临床实验数据显示,在症状出现3 d内接收该药物治疗的患者住院和死亡风险显著降低89%[23]。
-
在药物研发领域,拟肽类抑制剂虽然具有较高的生物活性等优点,但其开发仍面临着诸多挑战,例如合成难度大、成本高昂以及结构优化难度大等问题。相比之下,非拟肽类抑制剂具有分子量小,合成步骤简单,易于进行结构改造等优势,从而显示出良好的成药潜力[24]。
-
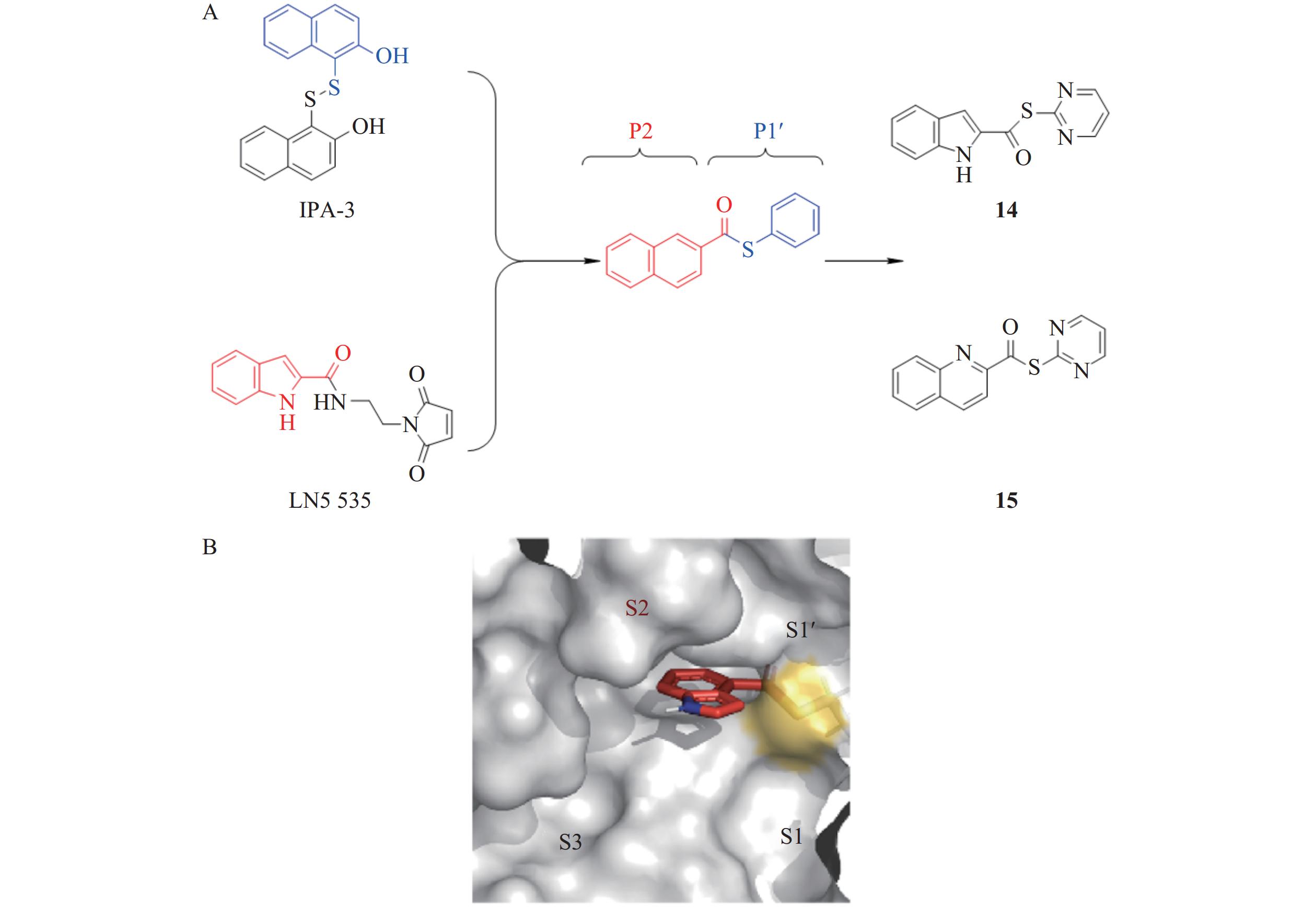
Pillaiyar等[25]通过计算机虚拟筛选和片段药物设计,发现IPA-3和LN5535两个活性片段分别结合在Mpro活性位点的P2和P1’结构域,对Mpro的抑制活性达到微摩尔级。在此基础上,研究人员通过片段连接和结构优化,设计并合成一系列新型硫酯类化合物,其中,化合物14和15展现出潜在的成药性价值(图6)。通过对Mpro与硫酯类化合物14、15的共晶结构解析发现,在蛋白酶Mpro催化裂解抑制剂的过程中,嘧啶硫醇盐(pyrimidine thiolate)作为离去基团离开了活性位点,而化合物的剩余片段与Cys145发生共价结合,形成硫酯型酶-抑制剂复合物。具有催化活性的残基His41和Met165与化合物14、15形成稳定的π-π相互作用,这种作用如同“钳夹”一般,牢牢捕获了抑制剂的芳香吲哚环(14)和喹啉环(15),从而实现与抑制剂的稳定结合。进一步研究表明,硫酯键是抑制剂发挥作用的必需基团,且抑制剂的离去基团必须为芳杂环。其中,以嘧啶环作为离去基团的化合物活性最佳,而离去基团为苯环的化合物则无活性。构效关系研究表明,化合物14表现出较强的广谱Mpro抑制活性,对SARS-CoV-2 Mpro的IC50值为11.4 nmol/L。在Calu-3和Vero76细胞模型中,化合物14显示出纳摩尔级别的抗病毒活性,EC50为0.11 µmol/L,且对宿主细胞无毒性。此外,该类抑制剂不仅对SARS-CoV-2 Mpro有效,还对SARS-CoV-1和MERS-CoV的Mpro表现出高抑制活性,表明它具有广谱抗冠状病毒的潜力[26]。
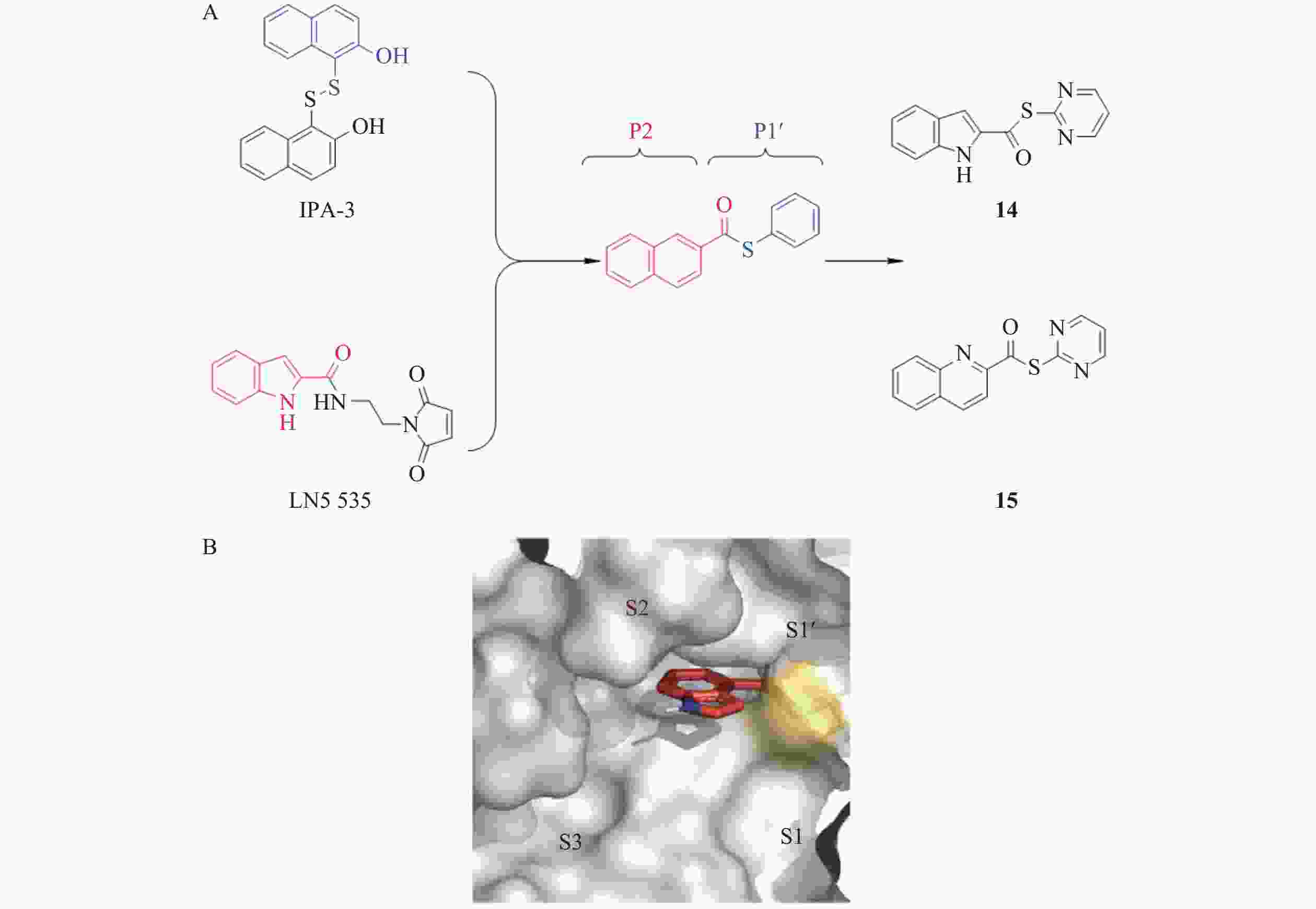
图 6 硫酯类抑制剂
-
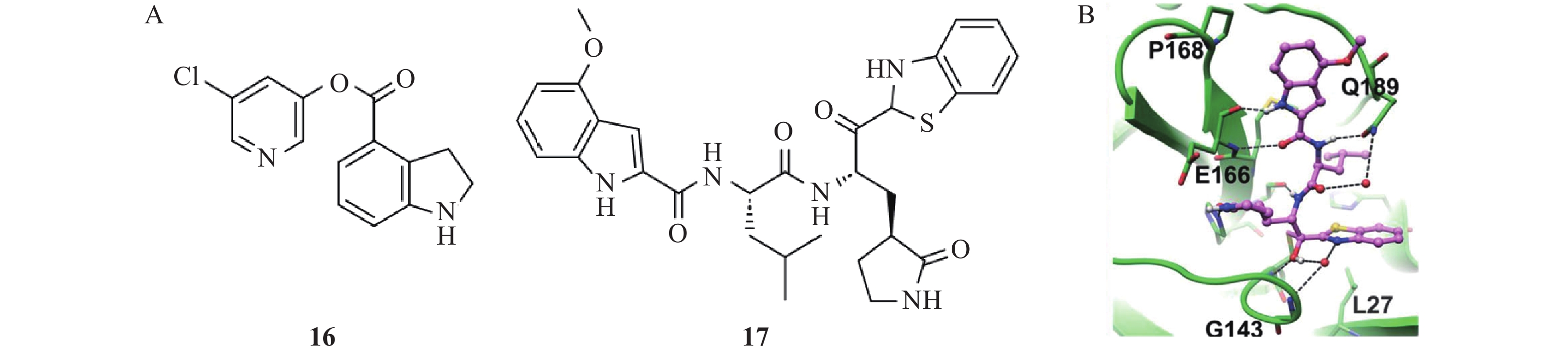
Hattori等[27]设计和合成的吲哚类化合物16和17均表现出显著的Mpro抑制活性(图7),但两者的作用机制存在差异。化合物16是一种不可逆的Mpro共价抑制剂,其作用机制涉及Mpro的催化残基Cys145、His41对化合物16酯碳发起亲核攻击,促使氯吡啶基酰化离去,同时羰基吲哚啉与Cys145通过共价键结合,从而抑制蛋白酶活性,其IC50为0.32 μmol/L,EC50为15 μmol/L。相比之下,化合物17是一种可逆的共价抑制剂,其苯并噻唑环邻位的羰基碳与Cys145的巯基发生亲核加成反应,形成可逆的共价键,该反应过程还伴随着羰基向醇的转化,不仅形成直接的氢键相互作用,还通过水分子介导形成广泛氢键相互作用网,进一步增强结合稳定性。晶体结构分析表明,化合物17中的4-甲氧基吲哚部分,被Pro168和Ala191环绕,中心部分朝向结合凹槽内部的Met165,这种空间位阻和疏水相互作用的匹配,使得化合物17能够较好地嵌入结合口袋中,增强了与Mpro的结合力[28]。与化合物16相比,化合物17的抗病毒活性更强,EC50为4.2 μmol/L。其通过与Mpro的多重相互作用实现了更强的结合,展现更优的酶抑制活性和抗病毒活性。此外,化合物17在高达200 μmol/L的浓度下未表现出显著的细胞毒性,而在同等浓度下瑞德西韦已显现毒性。进一步研究发现,化合物17与瑞德西韦联合使用对SARS-CoV-2具有协同抑制作用。免疫细胞化学实验证实,联合用药能完全阻断病毒感染,而单独使用时均有病毒突破现象,这一发现为开发联合治疗方案提供了重要依据。
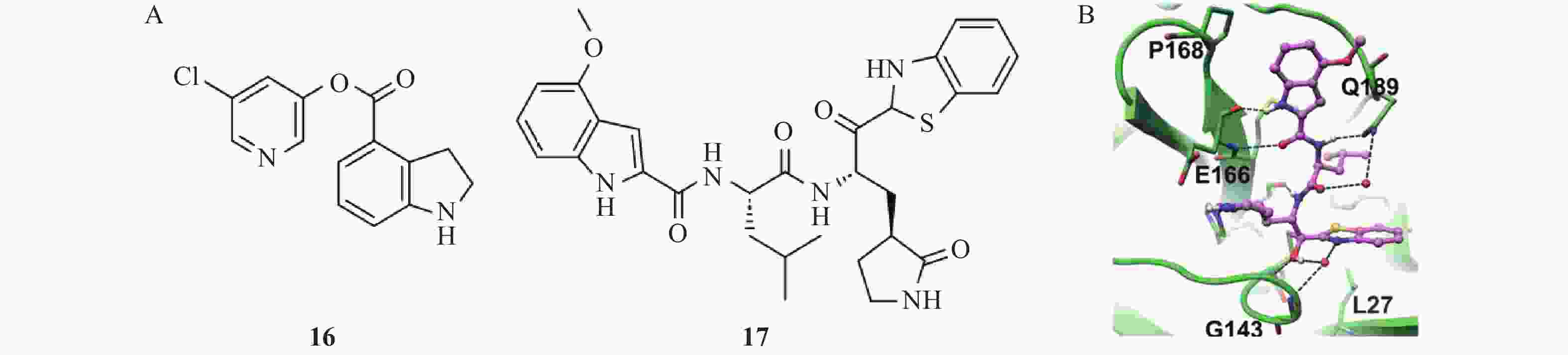
图 7 吲哚类共价抑制剂
-
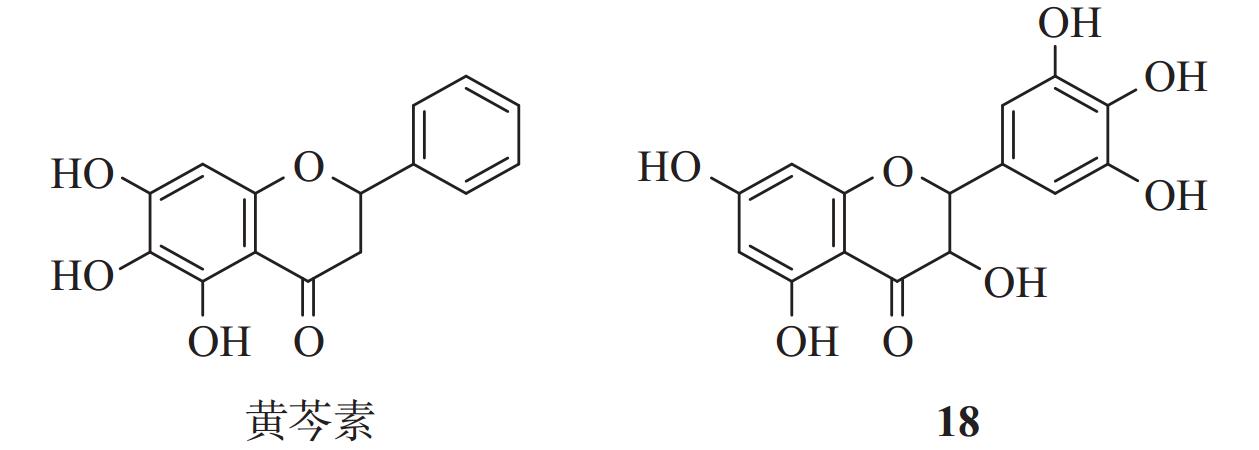
双黄连制剂是一种具有悠久历史的中成药,广泛用于治疗呼吸道感染。Liu等[29]通过FRET蛋白酶法发现双黄连制剂对SARS-CoV-2 Mpro具有抑制作用。进一步研究揭示,其活性成分黄芩素(图8)是一个非共价、非拟肽的Mpro抑制剂,具有很强的抗病毒活性,其IC50为6.41 μmol/L,EC50为1.69 μmol/L。机制研究表明,黄芩素的3个酚羟基与Mpro的Leu141和Gly143等残基侧链形成多个氢键相互作用,羰基与Glu166形成氢键相互作用。黄芩素游离苯环深入Mpro空腔,并与周围残基形成疏水相互作用[30]。此外,Cys145和His41还分别与黄芩素的芳环形成s-π和π-π相互作用。这些作用将黄芩素稳定在结合口袋的核心区域,并通过空间位阻阻止底物进入催化中心,从而实现对Mpro的活性抑制。
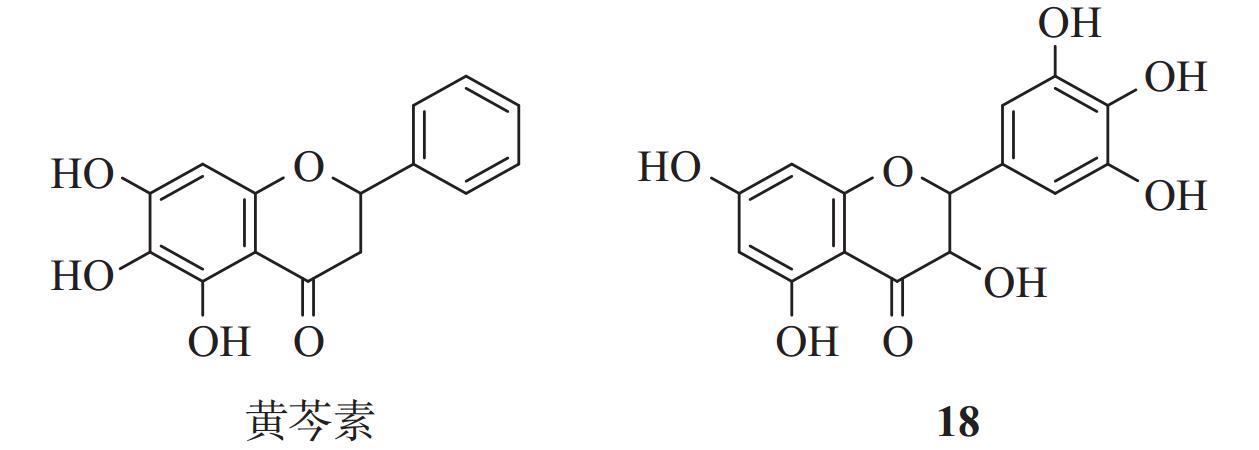
图 8 黄酮类抑制剂
通过FRET蛋白酶法,研究人员进一步筛选了一系列黄酮类似物,其中,化合物18表现出较好的抑制活性,IC50值为0.63 µmol/L,EC50值为8.0 µmol/L。与黄芩素不同,化合物18的联苯三酚结构不仅与Mpro催化残基形成共价结合,还通过苯酚的羟基与G143、S144、C145、T26等残基形成氢键,并与His41侧链形成了π-π相互作用[31]。联苯三酚结构展现出作为共价配体的潜力,这种独特的结合模式为新型Mpro抑制剂设计提供了新思路。
-
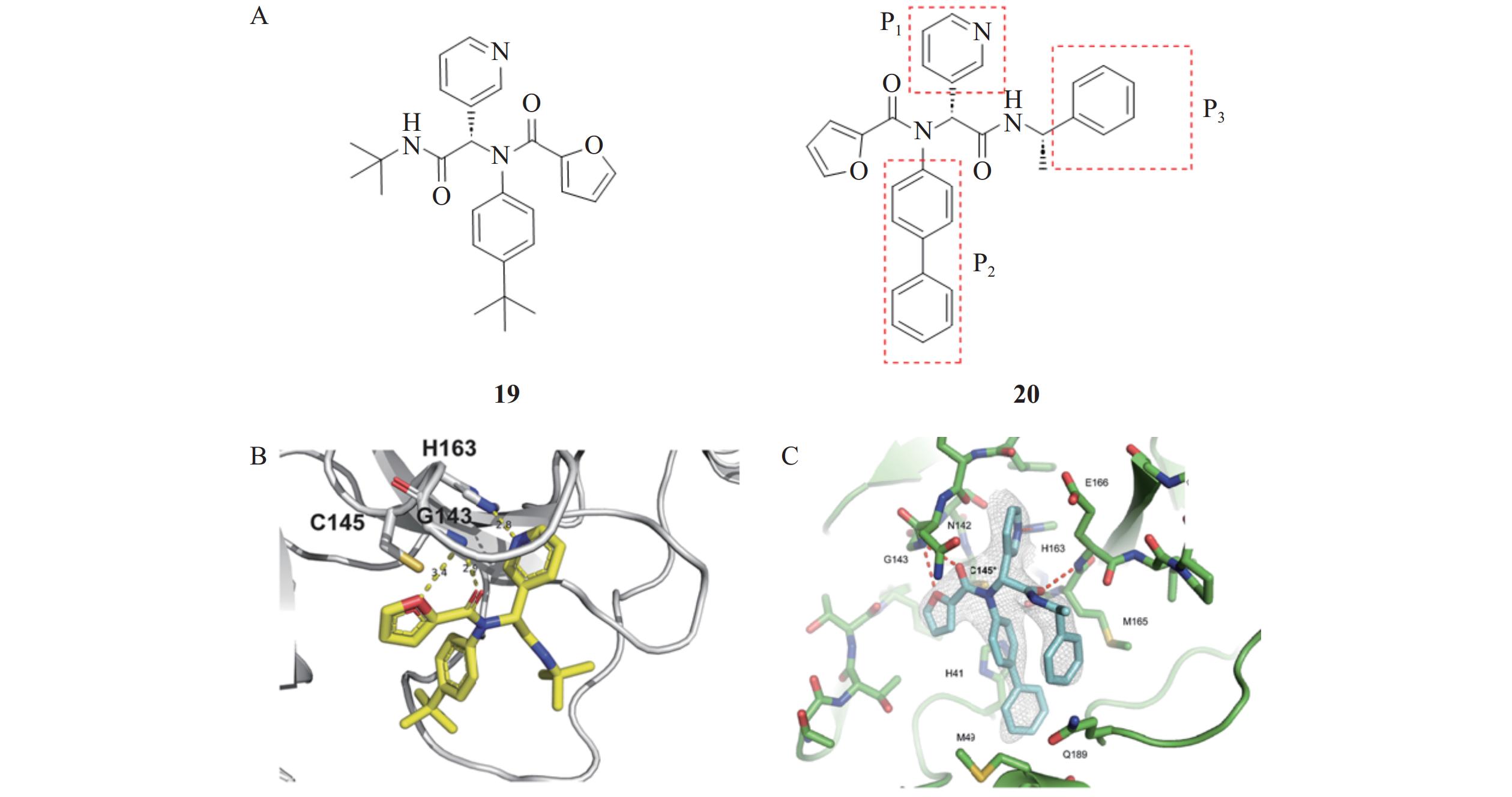
Lockbaum等[32]通过高通量筛选和结构优化发现了一种新型的呋喃酰胺类Mpro非共价抑制剂19(图9),基于FRET酶法测定其对SARS-CoV-2 Mpro的IC50为10.96 µmol/L,对SARS-CoV Mpro的IC50为11.23 µmol/L。晶体结构解析表明,化合物19的呋喃环能够深入Mpro的活性口袋,与关键氨基酸残基形成稳定的非共价相互作用,从而发挥抑制作用。Kitamura等[33]基于化合物19与Mpro的结合模式,利用Ugi四组分反应(U-4CR)方法,设计合成了呋喃酰胺类化合物20。在Vero E6细胞中,化合物20对SARS-CoV-2的EC50值为1.27 μmol/L,在人肺上皮Calu-3细胞中其对SARS-CoV-2的EC50值为3.03 μmol/L,展现出良好的抗病毒活性和成药潜力。晶体结构解析显示,化合物20的呋喃基与Mpro通过非共价相互作用结合。其中,P2位联苯插入Mpro的S2活性口袋,P1位的吡啶环占据S1口袋,并与His163形成氢键。连接吡啶环和α-苯甲基的酰胺键与Glu166主链形成氢键,且苯环部分位于S2和S4口袋之间。这种将苯基包围的结合形态导致分子内形成连续π-π堆积,增加了分子的稳定性,有助于增强化合物对Mpro的活性抑制[34]。
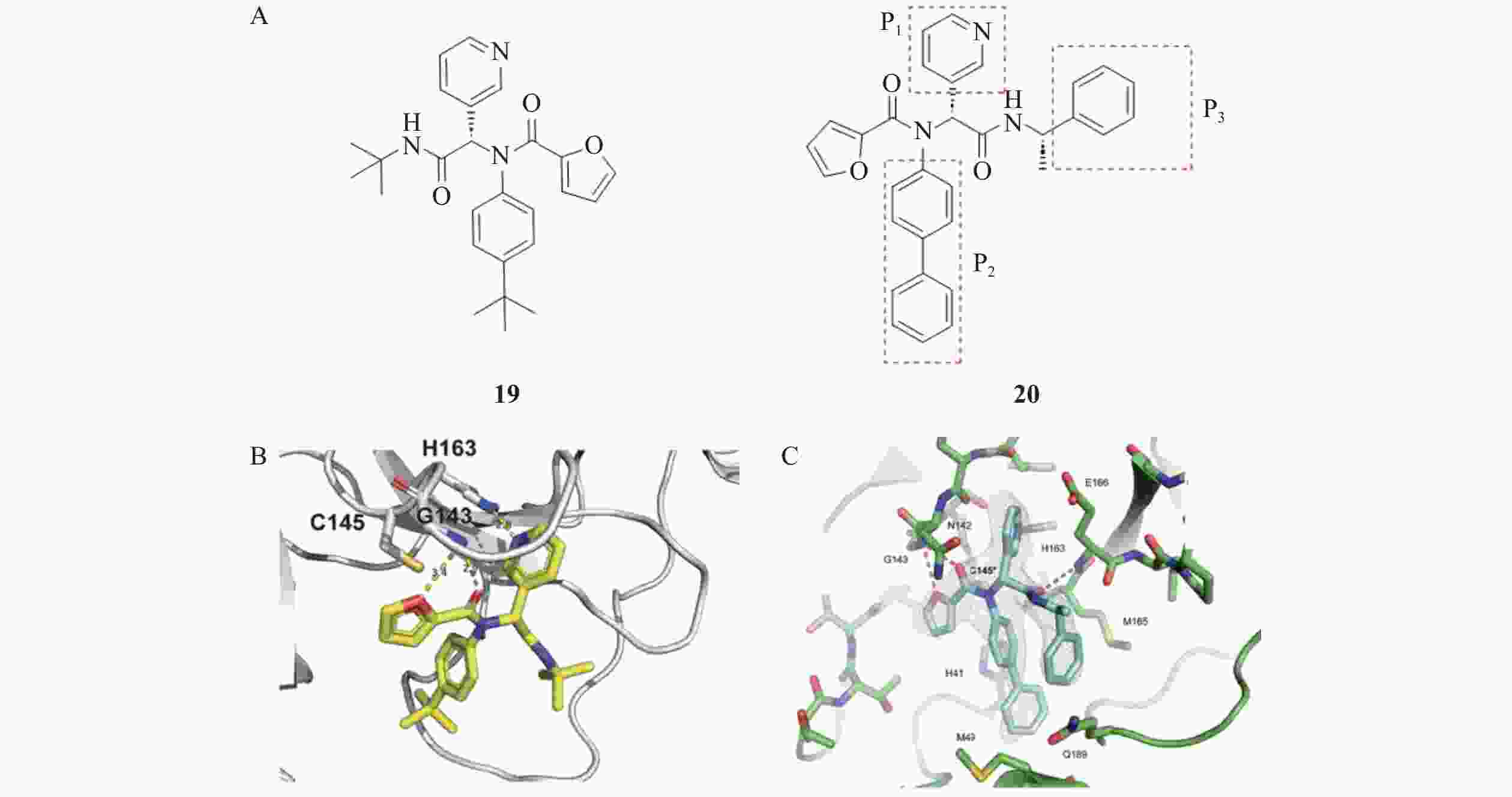
图 9 呋喃酰胺类抑制剂
构效关系研究发现,化合物20的P2取代基对抑制剂的活性和选择性有显著影响。其中,联苯基可能是Mpro口袋中可以容纳的最长取代基,完全占据了Mpro的疏水口袋,表现出最佳的酶抑制活性。将P3位置的取代基从叔丁基替换为α-甲基苄基后,显著提高了对酶的抑制活性。此外,α-甲基苄基的苯环部分结合于Mpro的口袋中间,这一全新的结合模式为Mpro抑制剂的设计提供了新的思路。
-
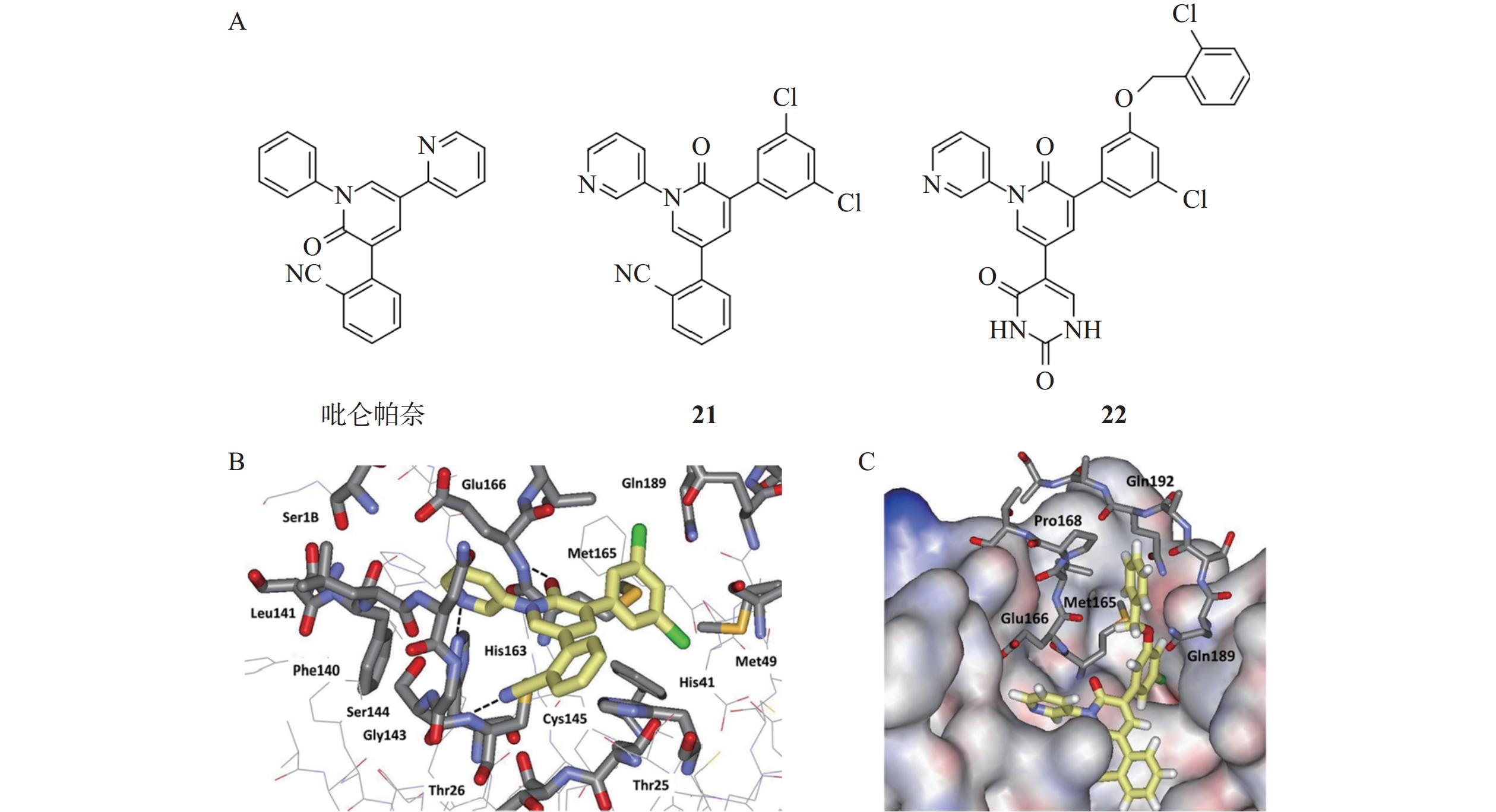
Zhang等[35]发现抗癫痫药吡仑帕奈具有Mpro抑制剂活性,IC50值为100 μmol/L(图10),随后通过自由能微扰(FEP)计算指导结构优化,设计并合成了一系列吡啶酮类抑制剂。其中,化合物21表现出中等以上的抑制活性,IC50值为4.0 μmol/L。晶体结构解析发现,化合物21与Mpro之间形成非共价相互作用,其吡啶酮氧原子、氰基氮原子和吡啶氮原子分别与Mpro活性位点Glu166、Cys145和His163形成氢键相互作用。化合物21的氯苯基边缘与Mpro S2口袋中His41的咪唑环紧密堆积,这种紧密的结合模式进一步增强其结合能力与稳定性。Mpro S3 - S4区域是底物结合口袋的一部分,从已有的晶体结构数据可知该区域存在一些疏水性氨基酸,例如,Met165、Leu167和Pro168等。通过向该区域引入能够与这些疏水性氨基酸发生相互作用的基团,可能会进一步提高化合物与Mpro的结合活性。因此,研究人员通过合理设计得到化合物22,在SARS-CoV-2 Mpro活性抑制方面表现出色,IC50值达到18 nmol/L,抗病毒活性EC50值为11.3 μmol/L,此外,化合物22具有良好的水溶性好,且未观察到明显的细胞毒性,展现出一定的成药潜力。
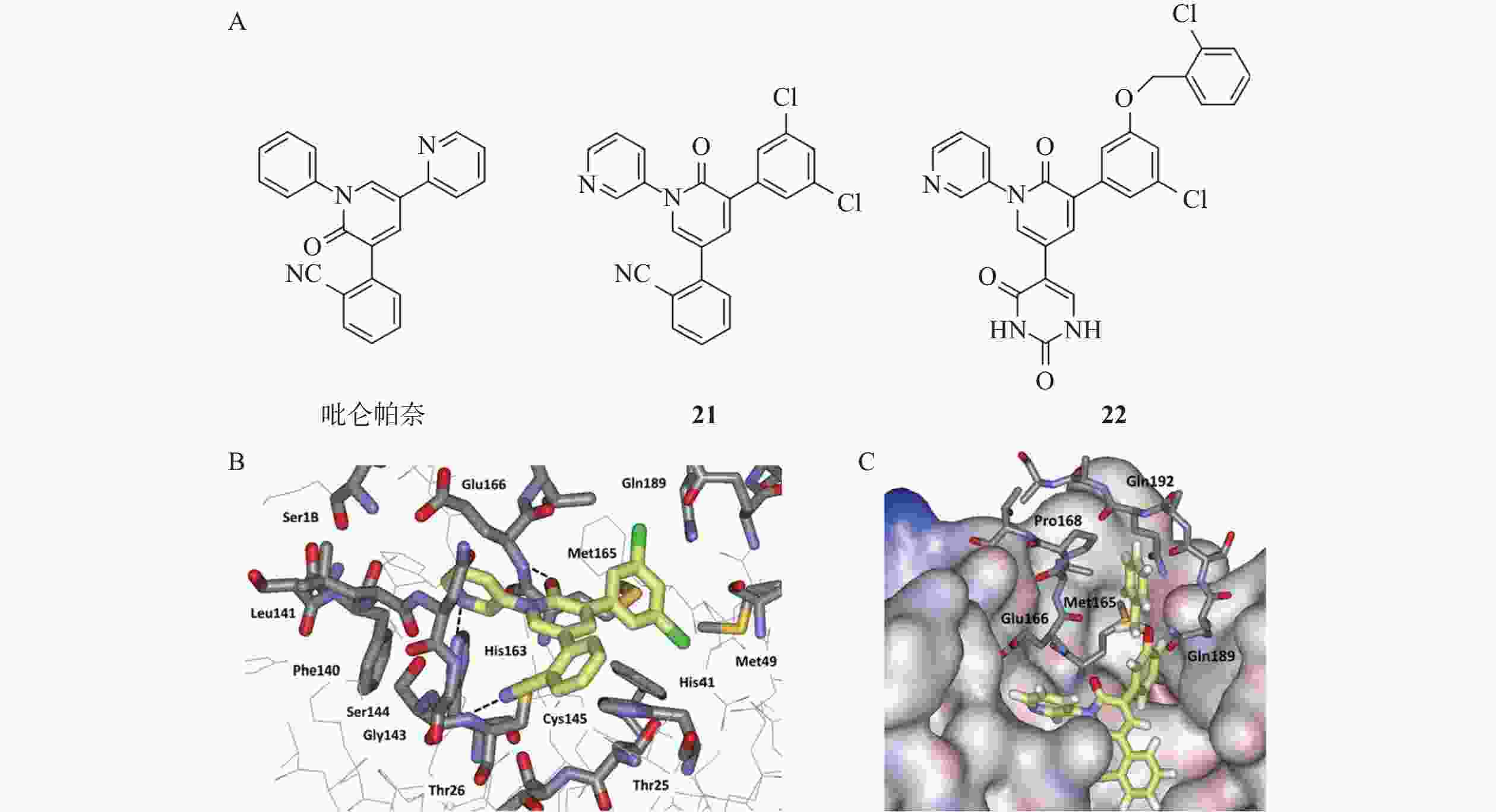
图 10 吡啶酮类抑制剂
-
PROTAC是药物研发领域的热点方向,通过特异性降解疾病相关蛋白,为多种疾病的治疗提供了全新的策略[36]。PROTAC是一种双功能分子,一端与靶蛋白结合,另一端与E3泛素连接酶结合,形成靶蛋白-PROTAC-E3三元复合物,从而劫持E3连接酶对靶蛋白进行泛素化修饰,随后被蛋白酶体识别并降解。与传统小分子抑制剂相比,PROTAC在提高药物选择性和作用效力、克服药物耐药性、降低毒副作用等方面具有显著优势[37-38]。在抗病毒药物研究领域,尤其是针对冠状病毒的研究中,研发Mpro的PROTAC降解剂具有较大前景。Mpro PROTAC降解剂能够直接清除病毒复制必需的关键蛋白酶Mpro,从而有效抑制病毒的复制和感染[39]。这种策略不仅有望能够提升药物作用效能,还能够克服因靶蛋白突变导致的耐药性问题,为抗冠状病毒药物的研发开辟了新的方向。
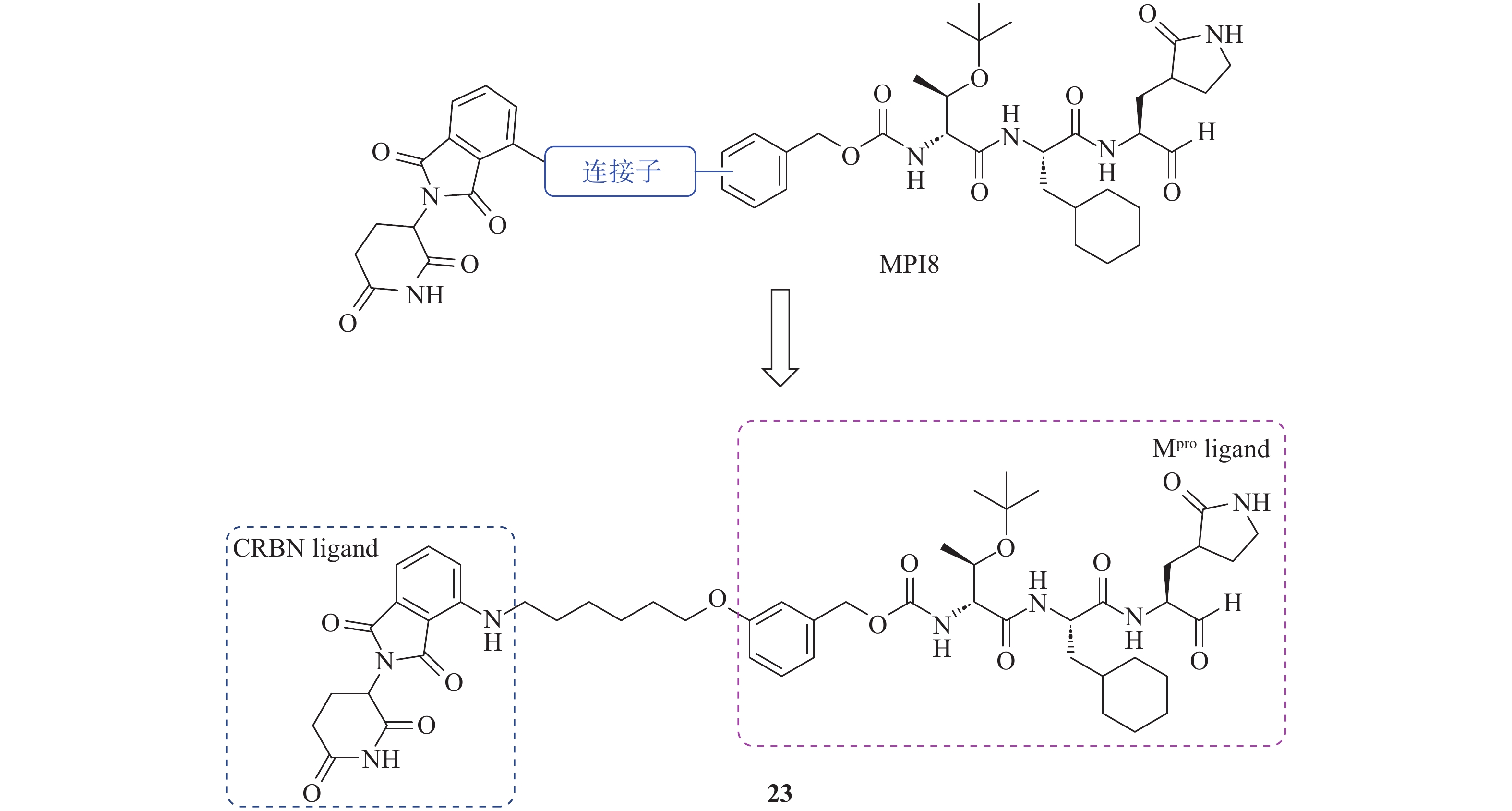
PROTAC分子由3个部分组成:靶蛋白配体、E3泛素连接酶配体以及连接两者的连接子(Linker)。Alugubelli等[40]和Nalawansha等[41]以抑制剂MPI8作为Mpro结合配体,泊马度胺作为E3连接酶CRBN(Cereblon)配体,设计并合成了针对Mpro的PROTAC分子(图11)。MPI8与Mpro结合模式显示,氮端的苯基指向溶剂区,为E3连接酶配体的连接位点提供了潜在的空间。研究发现,引入不同结构的连接子会显著影响PROTAC分子的空间构象,进而改变其与CRBN的结合亲和力及降解活性。当连接子为饱和烷烃时,随着碳链长度的增加,化合物对Mpro的抑制能力逐渐减弱;而降解活性则呈现先增加后下降的趋势,在连接子的碳原子数目为6时,化合物降解活性达到最佳。当连接子为PEG柔性链时,随着长度增加,化合物对Mpro的抑制能力逐渐下降且其抑制活性显著低于饱和烷烃类型的连接子。通过系统的构效关系研究,研究人员最终筛选得到化合物23,作用6 h即开始显著降解Mpro,12 h达到最大降解效果,DC50值为296 nmol/L。在A549-ACE2细胞中,化合物23对SARS-CoV-2表现出优秀的抗病毒活性,EC50值为492 nmol/L,且对先前流行的多种SARS-CoV-2毒株(包括WA.1、BA.1和XBB.1.5)均具有显著的抗病毒活性,在2.5 μmol/L浓度下降低病毒产量高达90%。此外,化合物23在克服耐药性上展现出显著优势,对含有NSP5 E166A突变(该突变使得重组SARS-CoV-2对奈玛特韦具有10倍抗性)的重组SARS-CoV-2病毒的抑制效果比对野生型病毒高5倍,为应对耐药性问题和开发广谱抗冠状病毒药物提供了候选分子。
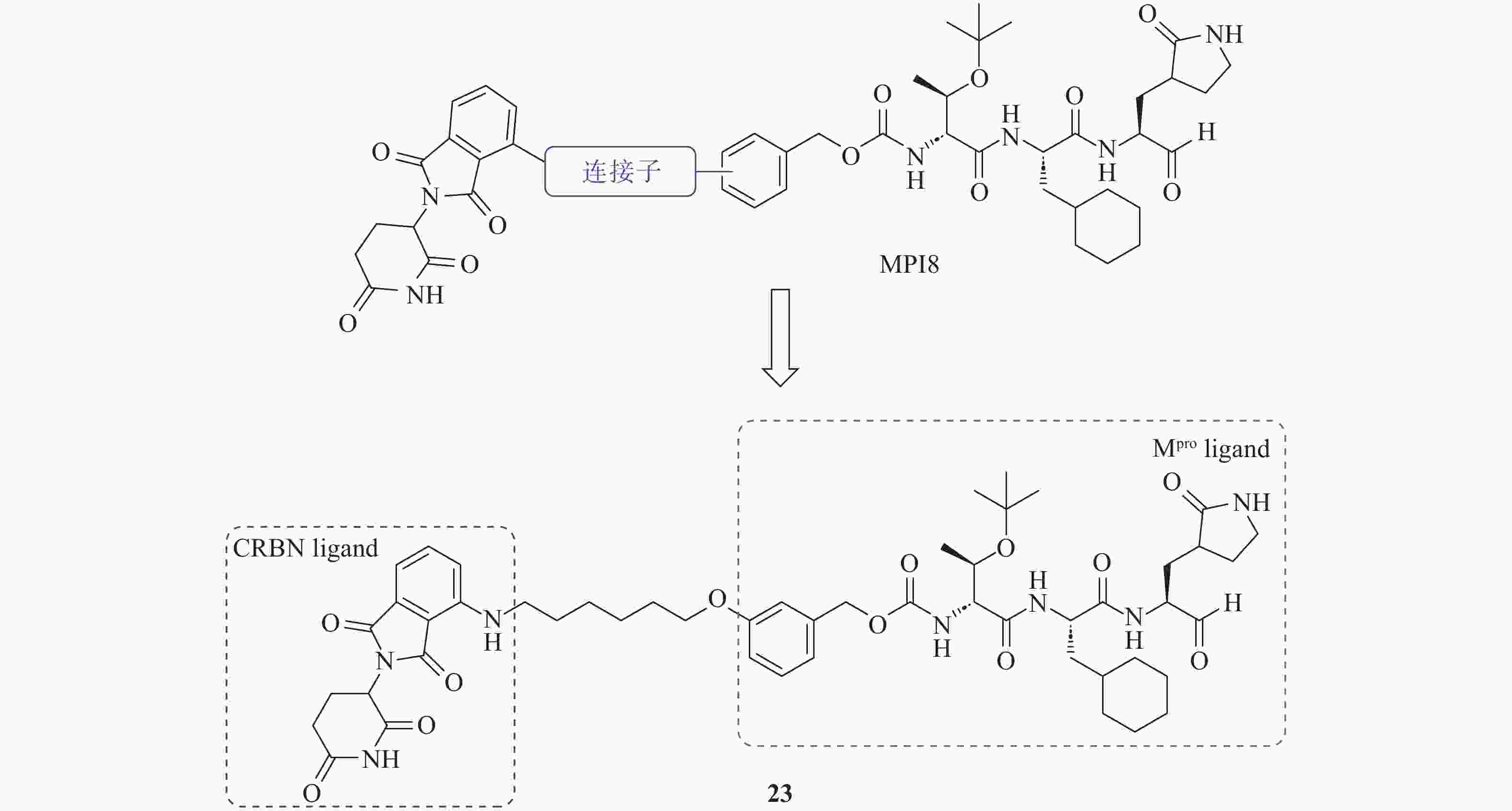
图 11 Mpro PROTAC 23的设计和结构
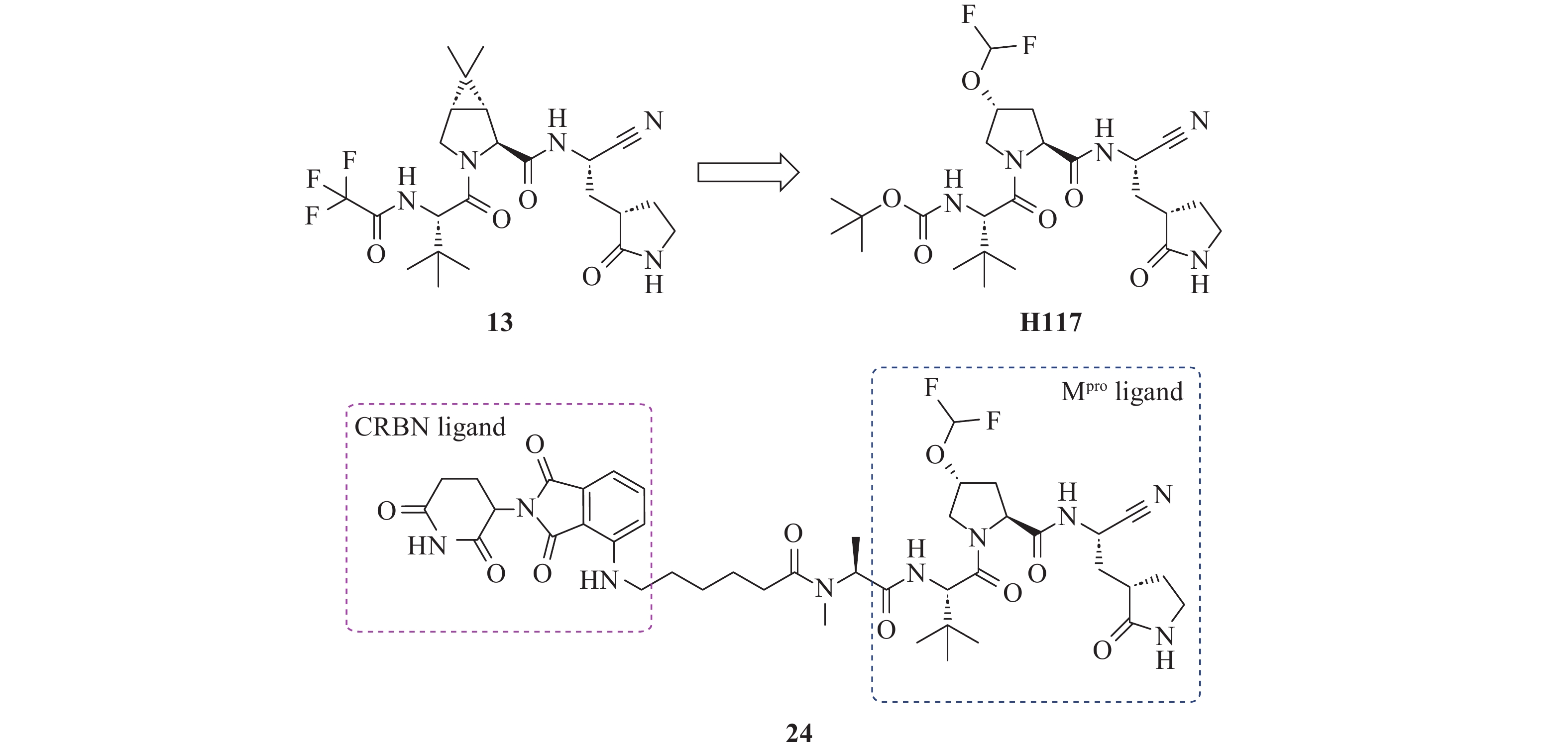
Sang等[42]利用PROTAC技术成功实现了对SARS-CoV-2 Mpro及其耐药突变体的有效清除。Mpro PROTAC的分子设计基于研究团队前期开发的Mpro抑制剂H117(IC50=151.3 nmol/L),其化学结构与临床上市药物奈玛特韦(13)相似(图12)。结合模式分析显示,H117的N-端叔丁氧羰基远离活性位点,适宜作为连接子连接位点。随后,研究人员通过N-端氨基将H117与CRBN配体泊马度胺偶联,并借鉴前期在HCV蛋白酶降解剂设计上的成功经验[43],选择长度为10个碳原子的饱和烷烃链作为连接子,连接H117和泊马度胺,从而合成了化合物24。酶抑制实验显示,化合物24对Mpro的抑制活性(IC50=181.9 nmol/L)与母体分子H117相当,表明连接子的引入未显著影响靶蛋白结合能力。在HEK-293T细胞中,化合物24以剂量依赖性方式高效降解Mpro(DC50=621 nmol/L),且在高达100 μmol/L浓度下未表现出细胞毒性。更重要的是,化合物24对耐药突变体E166V和E166A仍表现出显著降解能力,DC50值分别为589 nmol/L、602 nmol/L,展现了在应对病毒耐药性问题中的潜在优势。
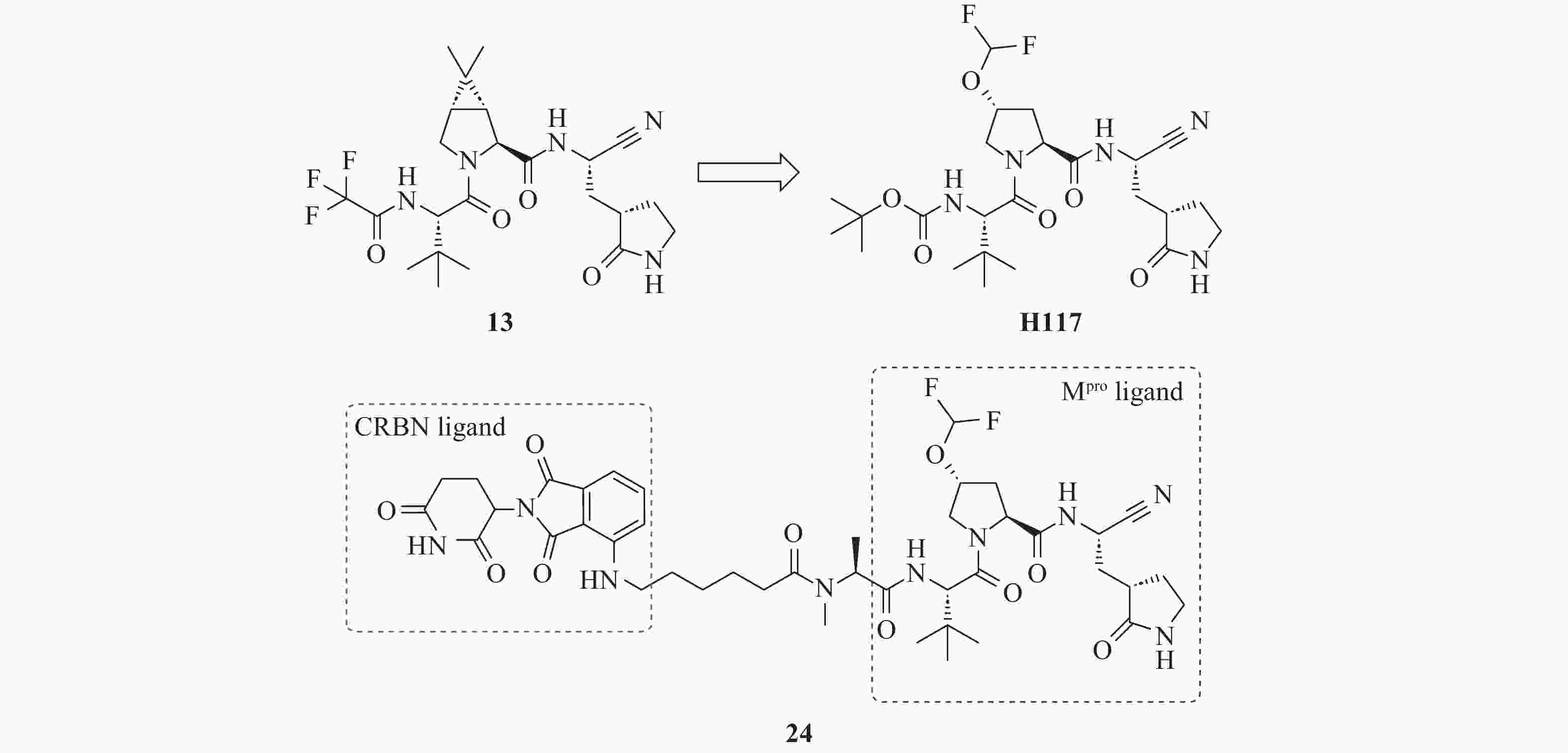
图 12 Mpro PROTAC 24的设计和结构
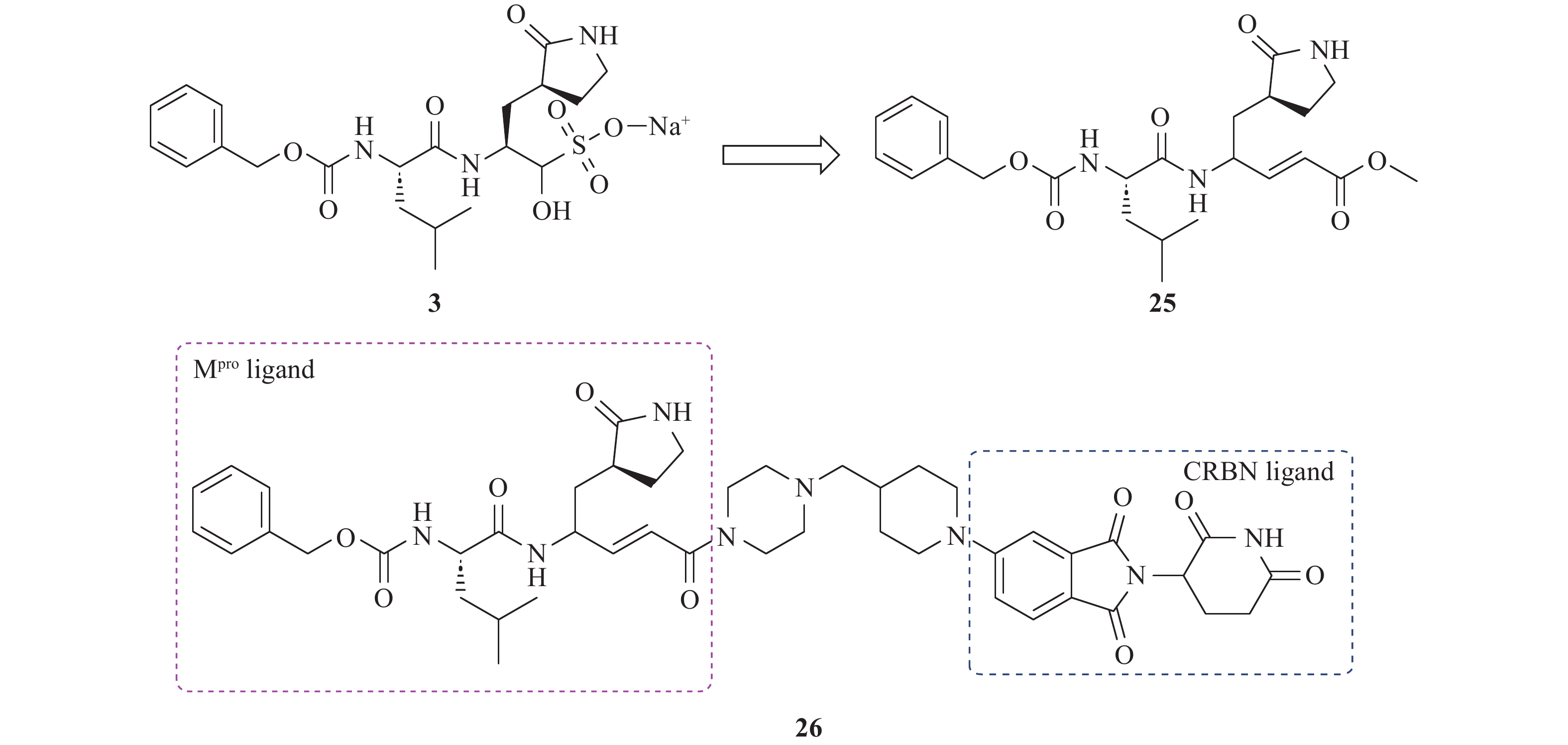
Gadd等[44]基于肽醛类化合物3进行Mpro PROTAC设计,将其醛基替换为α,β-不饱和甲酯,化合物25(图13),从而实现了与靶蛋白Mpro的可逆结合,确保了PROTAC分子在目标蛋白降解后能够解离并循环利用。这种可逆性不仅满足了高效降解目标蛋白的需求,还能够在较低剂量下实现更高的作用效率。此外,α,β-不饱和甲酯末端还可以作为E3连接酶配体的连接位点。研究人员参考Arvinas公司的PROTAC设计,以哌嗪-哌啶作为连接子,CRBN配体作为E3连接酶招募基团,设计并合成PROTAC分子。其中,化合物26对Mpro的IC50值为21.2 μmol/L,低于母体化合物,表明连接子引入可能影响了活性。在HeLa细胞中,化合物26以剂量依赖性方式降低Mpro水平,在10 μmol/L浓度下降解率达70%,且未表现出细胞毒性。然而,化合物26活性较低,且未在病毒感染模型中验证功能,限制了其临床转化潜力。
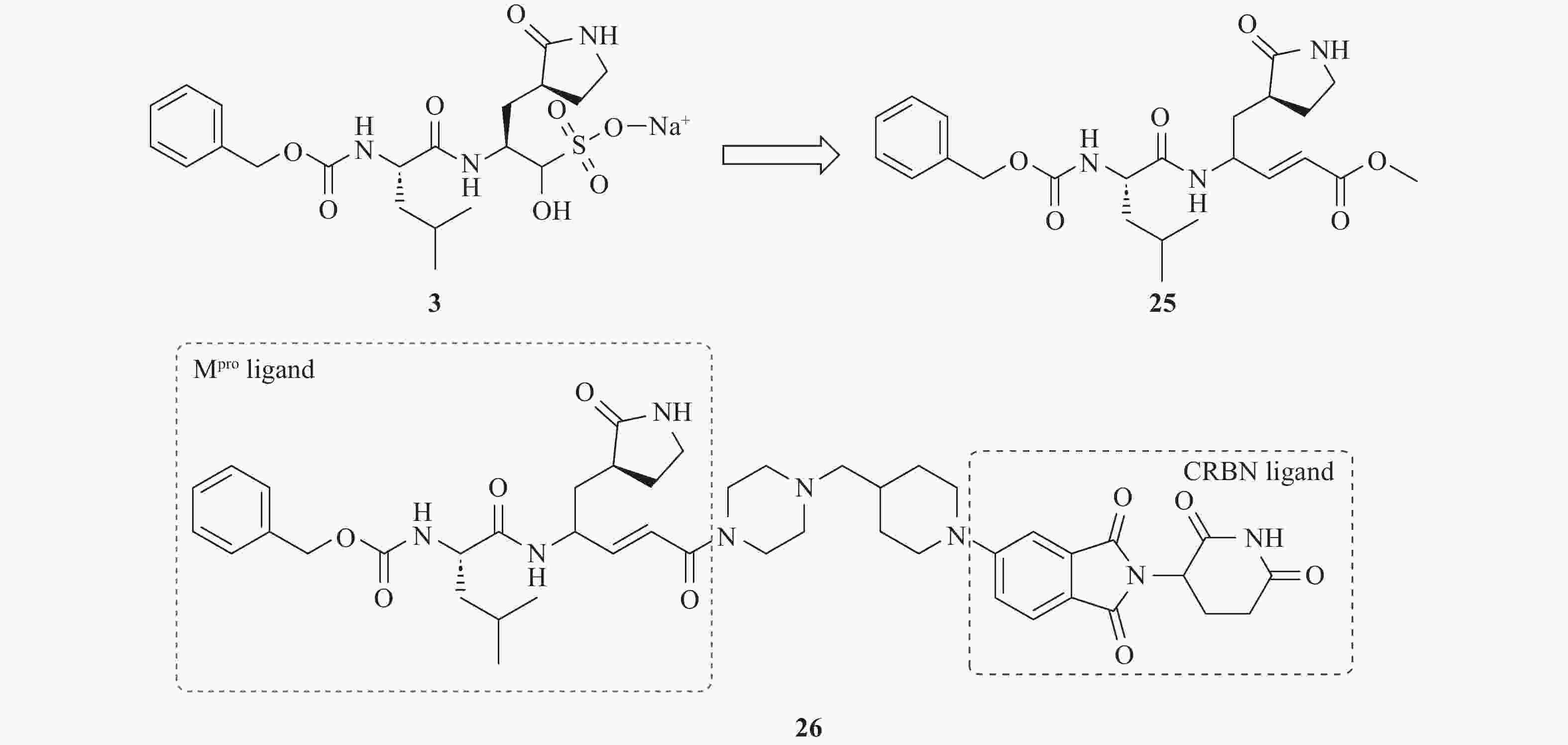
图 13 Mpro PROTAC 26的设计和结构
-
新冠疫情的全球大流行对世界公共卫生体系带来了极其严峻的挑战,也让人们切实感受到高传染性病毒的巨大威胁。SARS-CoV-2 Mpro作为冠状病毒复制和感染的关键酶,是抗病毒药物开发的核心靶点之一。目前已上市的Mpro抑制剂主要有辉瑞公司研发的化合物奈玛特韦和先声药业研发的先诺特韦。奈玛特韦的Ⅲ期临床试验表明,在症状出现3 d内服用,可使高危患者的死亡风险降低89%[23]。对Omicron(BA.1、BA.2等亚分支)等变异株均表现出良好抑制活性。此外,该药还对SARS-CoV、MERS-CoV等也有较好的抑制作用,具有广谱的抗病毒活性。先诺特韦是由先声药业设计的针对Omicron变异株的Mpro抑制剂。临床Ⅱ期、Ⅲ期研究表明,服用先诺特韦可以显著降低病毒载量,缩短症状持续时间及核酸转阴时间,对轻中度SARS-CoV-2成年患者具有显著疗效[45-46]。
本综述系统梳理了近年来靶向Mpro抑制剂和降解剂的研究进展,涵盖了拟肽类共价抑制剂、非拟肽类共价抑制剂、非共价抑制剂以及靶向Mpro PROTAC降解剂等多种策略,深入探讨化合物设计方法、作用机制、构效关系、优化方向及应用价值。
拟肽类抑制剂通过模拟底物结构与Mpro活性位点形成共价键,展现出高效的抑制活性。例如,氰基弹头类化合物11的IC50值低至13 nmol/L,充分体现了其在靶向共价抑制中的优势。然而,这类化合物通常面临合成复杂性和潜在毒性的问题,限制了其进一步开发。相比之下,非拟肽类小分子抑制剂凭借分子量小、合成简便等优势,在提高成药性方面展现了巨大潜力。例如,硫酯类化合物14在体外实验中表现出优异的Mpro抑制效果(IC50=11.4 nmol/L),为开发高效、低成本的抗病毒药物提供了新方向。非共价抑制剂则通过与Mpro活性位点形成氢键、疏水相互作用等非共价键发挥作用,避免了共价抑制可能带来的脱靶毒性问题。尽管其抑制活性通常低于共价抑制剂,但其更高的选择性和安全性使其成为重要的研究方向。奈玛特韦与先诺特韦的上市标志着新冠病毒临床治疗进入临床治疗阶段,二者对变异株的有效性及良好的安全性奠定了临床应用基础。此外,PROTAC技术的引入为抗病毒药物研发开辟了全新思路。通过特异性降解Mpro,PROTAC不仅能够克服传统抑制剂因靶点突变导致的耐药性问题,还具有催化特性,在亚化学计量下实现靶蛋白高效降解。例如,化合物23对耐药突变体E166A的抑制效果比野生型高出5倍,充分展现了其在应对耐药性挑战中的优势。
尽管目前针对SARS-CoV-2 Mpro的药物研究取得了显著进展,但仍存在诸多亟待解决的问题。首先,病毒的高度变异性可能导致现有抑制剂失效,因此需要开发广谱抗病毒药物以应对未来可能出现的新型变异株。其次,现有抑制剂在药代动力学特性、口服生物利用度、毒性和代谢稳定性等方面仍有较大优化空间。此外,PROTAC技术虽然前景广阔,但其在生物体内的降解效率、抗病毒药效及安全性仍需进一步验证。随着人工智能、计算化学和结构生物学等技术的快速发展,有望实现Mpro抑制剂精准地设计和快速优化。同时,PROTAC技术的成熟应用将为克服耐药性问题提供全新的解决方案。
Research progress on inhibitors and degraders of the main protease of SARS-CoV-2
-
摘要: 新型冠状病毒(SARS-CoV-2)主蛋白酶(Mpro)是病毒复制和转录的关键酶,因其高度保守性及与宿主蛋白酶的低同源性,成为抗病毒药物研发的重要靶点。本文综述近年来针对Mpro抑制剂包括拟肽类共价抑制剂、非拟肽类共价抑制剂和非共价类抑制剂的研究进展,深入探讨蛋白降解靶向嵌合体(PROTAC)技术在Mpro降解剂中的应用,为抗病毒药物的研发提供参考。Abstract: The main protease (Mpro) of SARS-CoV-2, a pivotal enzyme for viral replication and transcription, has emerged as a critical therapeutic target in antiviral drug discovery due to its high conservation across coronaviruses and low homology with host proteases. Recent advances in Mpro inhibitors were summarized in this review, including peptide-mimetic covalent inhibitors, non-peptide covalent inhibitors, and non-covalent inhibitors. Furthermore, the application of proteolysis targeting chimera (PROTAC) technology in developing Mpro degraders was explored, which provided valuable insights for the development of antiviral drugs.
-
[1] WALDMAN R H, GANGULY R. Immunity to infections on secretory surfaces[J]. J Infect Dis, 1974, 130(4):419-440. doi: 10.1093/infdis/130.4.419 [2] LAN J, GE J W, YU J F, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor[J]. Nature, 2020, 581(7807):215-220. doi: 10.1038/s41586-020-2180-5 [3] C E. WHO coronavirus disease(COVID-19)[M]. Weekly Epidemiological Update, 2021. [4] FEHR A R, PERLMAN S. Coronaviruses: an overview of their replication and pathogenesis[J]. Methods Mol Biol, 2015, 1282:1-23. [5] Shannon A, Selisko B, Le N, et al. Favipiravir strikes the SARS-CoV-2 at its Achilles heel, the RNA polymerase [J]. bioRxiv, 2020. [6] ZHOU P, YANG X L, WANG X G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin[J]. Nature, 2020, 579(7798):270-273. doi: 10.1038/s41586-020-2012-7 [7] FORNI D, CAGLIANI R, CLERICI M, et al. Molecular evolution of human coronavirus genomes[J]. Trends Microbiol, 2017, 25(1):35-48. doi: 10.1016/j.tim.2016.09.001 [8] LI G, HILGENFELD R, WHITLEY R, et al. Therapeutic strategies for COVID-19: progress and lessons learned[J]. Nat Rev Drug Discov, 2023, 22(6): 449-475. [9] CAPASSO C, NOCENTINI A, SUPURAN C T. Protease inhibitors targeting the main protease and papain-like protease of coronaviruses[J]. Expert Opin Ther Pat, 2021, 31(4):309-324. doi: 10.1080/13543776.2021.1857726 [10] KONNO S, KOBAYASHI K, SENDA M, et al. 3CL protease inhibitors with an electrophilic arylketone moiety as anti-SARS-CoV-2 agents[J]. J Med Chem, 2022, 65(4):2926-2939. doi: 10.1021/acs.jmedchem.1c00665 [11] ZHANG L L, LIN D Z, SUN X, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors[J]. Science, 2020, 368(6489):409-412. doi: 10.1126/science.abb3405 [12] ZHANG L L, LIN D Z, KUSOV Y, et al. α-ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment[J]. J Med Chem, 2020, 63(9):4562-4578. doi: 10.1021/acs.jmedchem.9b01828 [13] PERERA K D, RATHNAYAKE A D, LIU H W, et al. Characterization of amino acid substitutions in feline coronavirus 3C-like protease from a cat with feline infectious peritonitis treated with a protease inhibitor[J]. Vet Microbiol, 2019, 237:108398. doi: 10.1016/j.vetmic.2019.108398 [14] MA C L, SACCO M D, HURST B, et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease[J]. Cell Res, 2020, 30(8): 678-692. [15] YANG K S, MA X R, MA Y Y, et al. A quick route to multiple highly potent SARS-CoV-2 main protease inhibitors[J]. Chem Med Chem, 2021, 16(6):942-948. doi: 10.1002/cmdc.202000924 [16] GÜNTHER S, REINKE P Y A, FERNÁNDEZ-GARCÍA Y, et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease[J]. Science, 2021, 372(6542):642-646. doi: 10.1126/science.abf7945 [17] DAI W H , ZHANG B, JIANG X M, et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease[J]. Science, 2020, 368(6497):1331-1335. [18] De Vries M, Mohamed A S, Prescott R A, et al. A comparative analysis of SARS-CoV-2 antivirals characterizes 3CLpro inhibitor PF-00835231 as a potential new treatment for COVID-19 [J]. J Virol, 2021, 95(7): e01819-20. [19] HOFFMAN R L, KANIA R S, BROTHERS M A, et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19[J]. J Med Chem, 2020, 63(21):12725-12747. doi: 10.1021/acs.jmedchem.0c01063 [20] BORAS B, JONES R M, ANSON B J, et al. Discovery of a novel inhibitor of coronavirus 3CL protease for the potential treatment of COVID-19[J]. bioRxiv, 2021. [21] ZHAO Y, FANG C, ZHANG Q, et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332[J]. Protein Cell, 2022, 13(9):689-693. doi: 10.1007/s13238-021-00883-2 [22] BAI B, ARUTYUNOVA E, KHAN M B, et al. Peptidomimetic nitrile warheads as SARS-CoV-2 3CL protease inhibitors[J]. RSC Med Chem, 2021, 12(10):1722-1730. doi: 10.1039/D1MD00247C [23] OWEN D R, ALLERTON C M N, ANDERSON A S, et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19[J]. Science, 2021, 374(6575):1586-1593. doi: 10.1126/science.abl4784 [24] KREUTZER A G, KRUMBERGER M, DIESSNER E M, et al. A cyclic peptide inhibitor of the SARS-CoV-2 main protease[J]. Eur J Med Chem, 2021, 221:113530. doi: 10.1016/j.ejmech.2021.113530 [25] PILLAIYAR T, FLURY P, KRÜGER N, et al. Small-molecule thioesters as SARS-CoV-2 main protease inhibitors: enzyme inhibition, structure-activity relationships, antiviral activity, and X-ray structure determination[J]. J Med Chem, 2022, 65(13):9376-9395. doi: 10.1021/acs.jmedchem.2c00636 [26] FERREIRA G M, KRONENBERGER T, TONDURU A K, et al. SARS-COV-2 Mpro conformational changes induced by covalently bound ligands[J]. J Biomol Struct Dyn, 2022, 40(22):12347-12357. doi: 10.1080/07391102.2021.1970626 [27] HATTORI S I, HIGASHI-KUWATA N, HAYASHI H, et al. A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication[J]. Nat Commun, 2021, 12(1):668. doi: 10.1038/s41467-021-20900-6 [28] Hattori S I, Higshi-Kuwata N, Raghavaiah J, et al. GRL-0920, an Indole Chloropyridinyl Ester, Completely Blocks SARS-CoV-2 Infection [J]. mBio, 2020, 11(4): e01833-20. [29] LIU H B, YE F, SUN Q, et al. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro[J]. J Enzyme Inhib Med Chem, 2021, 36(1):497-503. doi: 10.1080/14756366.2021.1873977 [30] ZONG K, WEI C, LI W, et al. Identification of novel SARS-CoV-2 3CLpro inhibitors by molecular docking, in vitro assays, molecular dynamics simulations and DFT analyses[J]. Front Pharmacol, 2024, 15:1494953. doi: 10.3389/fphar.2024.1494953 [31] SU H X, YAO S, ZHAO W F, et al. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients[J]. Acta Pharmacol Sin, 2020, 41(9):1167-1177. doi: 10.1038/s41401-020-0483-6 [32] Lockbaum G J, Reyes A C, Lee J M, et al. Crystal Structure of SARS-CoV-2 Main Protease in Complex with the Non-Covalent Inhibitor ML188 [J]. Viruses, 2021, 13(2): 174. [33] KITAMURA N, SACCO M D, MA C L, et al. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors[J]. J Med Chem, 2022, 65(4):2848-2865. doi: 10.1021/acs.jmedchem.1c00509 [34] TURLINGTON M, CHUN A, TOMAR S, et al. Discovery of N-(benzo [1, 2, 3] triazol-1-yl)-N-(benzyl)acetamido)phenyl)carboxamides as severe acute respiratory syndrome coronavirus(SARS-CoV)3CLpro inhibitors: identification of ML300 and noncovalent nanomolar inhibitors with an induced-fit binding[J]. Bioorg Med Chem Lett, 2013, 23(22):6172-6177. doi: 10.1016/j.bmcl.2013.08.112 [35] ZHANG C H, STONE E A, DESHMUKH M, et al. Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations[J]. ACS Cent Sci, 2021, 7(3):467-475. doi: 10.1021/acscentsci.1c00039 [36] HSU J T, KUO C J, HSIEH H P, et al. Evaluation of metal-conjugated compounds as inhibitors of 3CL protease of SARS-CoV[J]. FEBS Lett, 2004, 574(1-3):116-120. doi: 10.1016/j.febslet.2004.08.015 [37] BURSLEM G M, CREWS C M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery[J]. Cell, 2020, 181(1):102-114. doi: 10.1016/j.cell.2019.11.031 [38] KONSTANTINIDOU M, LI J Y, ZHANG B D, et al. PROTACs- a game-changing technology[J]. Expert Opin Drug Discov, 2019, 14(12):1255-1268. doi: 10.1080/17460441.2019.1659242 [39] YANG K S, LEEUWON S Z, XU S Q, et al. Evolutionary and structural insights about potential SARS-CoV-2 evasion of nirmatrelvir[J]. J Med Chem, 2022, 65(13):8686-8698. doi: 10.1021/acs.jmedchem.2c00404 [40] ALUGUBELLI Y R, XIAO J, KHATUA K, et al. Discovery of first-in-class PROTAC degraders of SARS-CoV-2 main protease[J]. J Med Chem, 2024, 67(8):6495-6507. doi: 10.1021/acs.jmedchem.3c02416 [41] NALAWANSHA D A, CREWS C M. PROTACs: an emerging therapeutic modality in precision medicine[J]. Cell Chem Biol, 2020, 27(8):998-1014. doi: 10.1016/j.chembiol.2020.07.020 [42] SANG X H, WANG J, ZHOU J, et al. A chemical strategy for the degradation of the main protease of SARS-CoV-2 in cells[J]. J Am Chem Soc, 2023, 145(50):27248-27253. doi: 10.1021/jacs.3c12678 [43] PAIVA S L, CREWS C M. Targeted protein degradation: elements of PROTAC design[J]. Curr Opin Chem Biol, 2019, 50:111-119. doi: 10.1016/j.cbpa.2019.02.022 [44] GADD M S, TESTA A, LUCAS X, et al. Structural basis of PROTAC cooperative recognition for selective protein degradation[J]. Nat Chem Biol, 2017, 13(5):514-521. doi: 10.1038/nchembio.2329 [45] JIANG X R, SU H X, SHANG W J, et al. Structure-based development and preclinical evaluation of the SARS-CoV-2 3C-like protease inhibitor simnotrelvir[J]. Nat Commun, 2023, 14(1):6463. doi: 10.1038/s41467-023-42102-y [46] AMPORNDANAI K, MENG X L, SHANG W J, et al. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives[J]. Nat Commun, 2021, 12(1):3061. doi: 10.1038/s41467-021-23313-7 -

 点击查看大图
点击查看大图
图(13)
计量
- 文章访问数: 9904
- HTML全文浏览量: 4522
- PDF下载量: 20
- 被引次数: 0
