

 下载:
下载:

-
肿瘤患者由于免疫功能低下容易发生感染,因此,侵袭性真菌感染(invasive fungal infections,IFIs)是肿瘤治疗过程中一种非常严重的并发症,在临床上或将成为人类健康的重大威胁。为此,寻找新型抗真菌、抗肿瘤效果一体化的小分子化合物是一种解决当前问题的有效策略[1]。
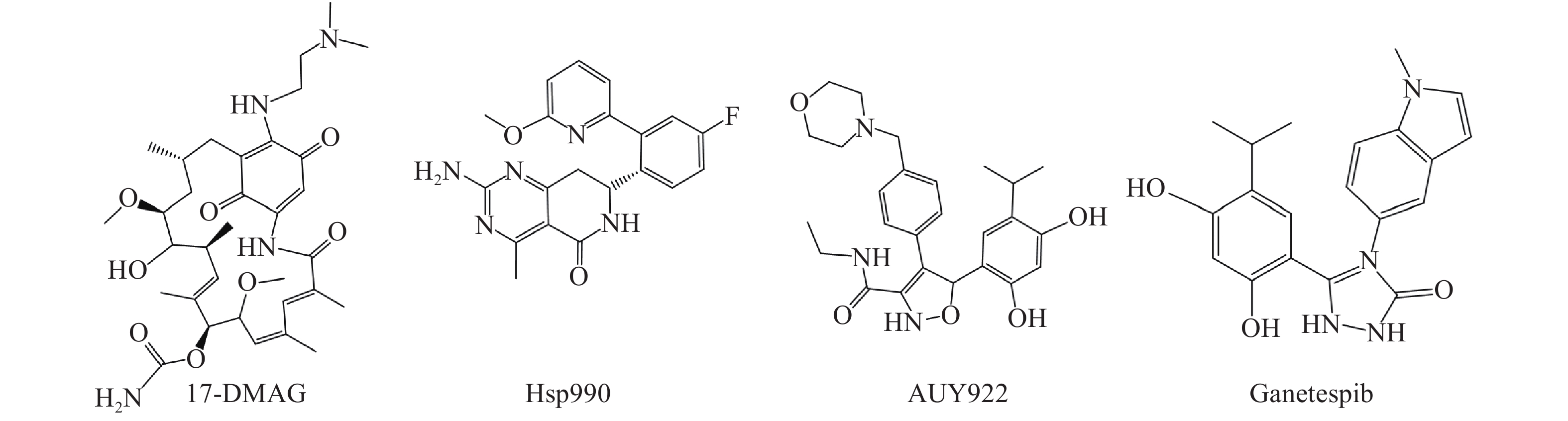
Hsp90是热休克蛋白家族中重要的分子伴侣,其通过与客户蛋白结合,可协助客户蛋白进行正确的折叠与组装,从而影响客户蛋白构象与功能[2]。许多研究报道表明,肿瘤细胞中Hsp90高表达,且它的众多客户蛋白如表皮生长因子受体(EGFR)、人表皮生长因子受体-2(Her2)和间变性淋巴瘤激酶(ALK)等也与肿瘤的增殖和转移等密切相关[3, 4]。因此,当前Hsp90抑制剂主要应用于抗肿瘤研究中,且已有多种结构类型的Hsp90抑制剂进入临床试验用于肿瘤的治疗,如17-DMAG、Hsp990、Ganetespib和AUY922等(图1)。
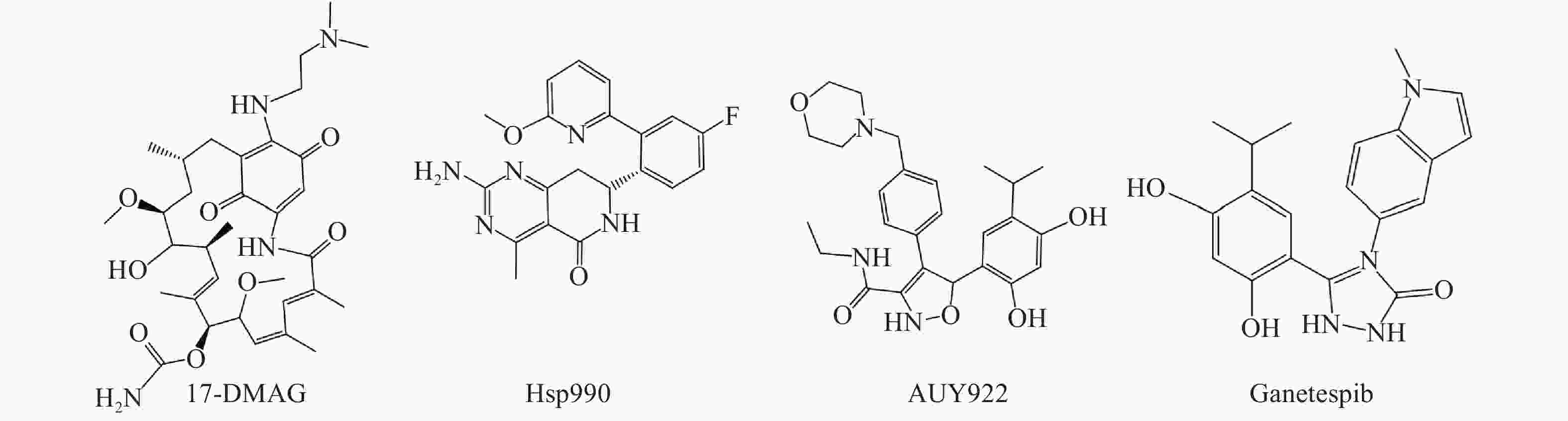
图 1 部分处于临床试验阶段的Hsp90抑制剂结构
真菌细胞压力应激反应的调节与真菌抗性密不可分,真菌中Hsp90调控应激反应,在多个耐药形成的信号通路中起到重要作用,抑制真菌Hsp90也可使耐药真菌恢复对抗真菌药物的敏感性[5]。此外作为可以诱导白念珠菌形态转变的热传感器,Hsp90参与调节真菌多个重要毒力因子的形成,如白念珠菌的生物被膜形成、菌丝态与酵母态之间的形态转换以及新生隐球菌荚膜的形成、黑素的产生等[6]。据报道,白念珠菌Hsp90核苷酸结合结构域与人源Hsp90相似。因此,以人源Hsp90抑制剂为先导化合物发现新型Hsp90抑制剂可能是获得兼具有抗耐药真菌和抗肿瘤双重作用的化合物的一个新思路。
Ganetespib是一种含有间二酚结构的第二代Hsp90抑制剂,其通过竞争性地与Hsp90的N端ATP结合位点结合,从而影响Hsp90生物学功能。当前Ganetespib针对多种类型的肿瘤已处于临床试验阶段,如非小细胞肺癌、前列腺癌、乳腺癌以及急性髓细胞白血病等。课题组前期研究表明,Ganetespib具有优秀的体外协同抗耐药真菌作用(FICI范围:0.023~0.039)[7],其与氟康唑(FLC)联用在感染耐药菌的ICR小鼠模型中显著降低肾脏荷菌量,并显著下调ERG11和外排泵基因CDR1、CDR2和MDR1的表达水平,初步探索了Ganetespib的抗耐药真菌机制[8]。
因此,本研究选择Ganetespib作为先导化合物开展进一步化合物的设计合成、协同抗耐药真菌和抗肿瘤活性及其作用机制研究,为研发一种兼具抗肿瘤作用的新型抗耐药真菌增强剂来治疗耐药念珠菌感染。
-
HZ-2111K-B Shaking Incubator(太仓华利达实验设备公司);SW-CT-IF型单人双面超净化工作台(苏州安泰空气技术有限公司);HPX-160BSH-Ⅲ恒温恒湿箱(上海新苗医疗器械制造有限公司);XW-80A漩涡混合器(上海琪特分析仪器有限公司);infinite M200多功能酶标仪(Tecan Austria GmbH)。
-
甲醇、乙醇、石油醚、四氯化碳、水合肼、乙酸乙酯,CDCl3、DMSO-d6,合成工作底物详见实验方法。氢氧化钠(NaOH)、氯化钠(NaCl)、吗啡啉丙磺酸(MOPS)、乙醇、正己烷、环己烷、酵母浸膏、葡萄糖、蛋白胨等。
-
无特别提及柱层析以H型硅胶(中国青岛海洋化学)为固定相,并使用快速液相制备色谱仪(TELEDYNE ISCO, CombiFlash@Rf+)进行分离。薄层色谱以硅胶GF254为固定相。化合物的核磁共振谱由Bruker公司MSL-600型或MSL-300型核磁共振仪测试完成,测试时以四甲基硅烷为内标。核磁数据中化学位移和耦合常数单位分别为ppm和Hz,以s, bs, d, t, q, dd, m分别代表单峰、宽单峰、二重峰、三重峰、四重峰、双二重峰和多重峰。高分辨质谱由API-3000LC-MS质谱仪测试完成。所有目标化合物的纯度(均> 95%)由Agilent 1260型高效液相色谱仪测试完成,并以C18柱为固定相,CH3OH/H2O(8/2, V/V)为流动相。
-
以化合物29a为原料制备中间体32a。1H NMR(300 MHz, CDCl3)δ 10.04(s, 1H), 7.42-7.27(m, 12H), 7.13-7.06(m, 2H), 7.05-6.92(m, 2H), 6.29(s, 1H), 4.92(s, 2H), 4.63(s, 2H), 3.36-3.20(m, 1H), 1.20(s, 3H), 1.18(s, 3H)。
取中间体32a(0.400 g, 0.814 mmol, 1.0 equiv)溶解于无水甲醇(20 ml)中,向其中加入10%的Pd/C(0.043 g, 40.7 μmol, 0.05 equiv),在H2条件下室温搅拌反应12 h,过滤,滤液经减压除去溶剂,得残余物以流动相(CH2Cl2/CH3OH, 100/1-100/5, V/V)在硅胶柱层析中纯化得淡灰色固体状目标化合物F1(0.210 g,收率83%)。1H NMR(600 MHz, DMSO-d6) δ 11.94(s, 1H), 9.60(s, 1H), 9.38(s, 1H), 7.40-7.34(m, 2H), 7.33-7.29(m, 1H), 7.21-7.11(m, 2H), 6.83(s, 1H), 6.25(s, 1H), 3.04-2.94(m, 1H), 0.98(s, 3H), 0.97(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 157.43, 155.04, 154.60, 145.69, 134.78, 129.17(s, 2C), 128.05, 127.93, 127.04(s, 2C), 125.92, 105.16, 102.79, 25.96, 22.95(s, 2C). HRMS(ESI)m/z: calcd for C17H16N3O3 [M-H]−
310.1191 , Found310.1197 。 -
目标化合物的制备方法参考化合物F1。
淡灰色固体(0.199 g,收率76%)。1H NMR(600 MHz, DMSO-d6)δ 11.89(s, 1H), 9.62(s, 1H), 9.42(s, 1H), 7.11(d, J=8.9 Hz, 2H), 6.92(d, J=8.9 Hz, 2H), 6.81(s, 1H), 6.27(s, 1H), 3.73(s, 3H), 3.05-2.93(m, 1H), 0.99(s, 3H), 0.97(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 159.01, 157.38, 155.13, 154.83, 145.97, 128.56(s, 2C), 127.83, 127.41, 125.85, 114.44(s, 2C), 105.13, 102.82, 55.78, 25.94, 22.94(s, 2C)。
-
目标化合物的制备方法参考化合物F1。
淡灰色固体(0.223 g,收率85%)。1H NMR(600 MHz, DMSO-d6)δ 11.93(s, 1H), 9.62(s, 1H), 9.43(s, 1H), 7.27(t, J=8.1 Hz, 1H), 6.89-6.87(m, 1H), 6.86(s, 1H), 6.80-6.77(m, 1H), 6.75-6.73(m, 1H), 6.27(s, 1H), 3.63(s, 3H), 3.03-2.95(m, 1H), 1.00(s, 3H), 0.99(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 159.70, 157.44, 155.11, 154.47, 145.67, 135.77, 129.89, 127.96, 125.96, 119.08, 113.84, 112.60, 105.30, 102.77, 55.58, 25.96, 22.98(s, 2C). HRMS(ESI)m/z: calcd for C18H18N3O4 [M-H]−
340.1303 , Found 340.1391. -
目标化合物的制备方法参考化合物F1。
淡灰色固体(0.213 g,收率81%)。1H NMR(600 MHz, DMSO-d6)δ 11.89(s, 1H), 9.61(s, 1H), 9.53(s, 1H), 7.36(t, J=7.9 Hz, 1H), 7.24(d, J=7.9 Hz, 1H), 7.06(d, J=7.9 Hz, 1H), 6.98(t, J=7.9 Hz, 1H), 6.65(s, 1H), 6.27(s, 1H), 3.59(s, 3H), 2.99-2.87(m, 1H), 0.86(d, J=6.9 Hz, 3H), 0.84(d, J=6.9 Hz, 3H). 13C NMR(15 1 MHz, DMSO-d6)δ 157.15, 155.53, 155.32, 154.72, 146.49, 130.94, 130.54, 126.32, 125.65, 123.06, 120.99, 112.76, 104.72, 102.80, 56.07, 25.62, 22.95, 22.87。
-
目标化合物的制备方法参考化合物F1。
淡灰色固体(0.197 g,收率77%)。1H NMR(600 MHz, DMSO-d6)δ 11.89(s, 1H), 9.58(s, 1H), 9.37(s, 1H), 7.14(d, J=8.2 Hz, 2H), 7.04(d, J=8.2 Hz, 2H), 6.81(s, 1H), 6.24(s, 1H), 3.13-2.82(m, 1H), 2.25(s, 3H), 0.97(s, 3H), 0.96(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 157.42, 155.06, 154.69, 145.86, 137.55, 132.20, 129.64(s, 2C), 127.93, 126.91(s, 2C), 125.89, 105.23, 102.80, 25.98, 22.91(s, 2C), 21.02. HRMS(ESI)m/z: calcd for C18H18N3O3 [M-H]−
324.1354 , Found324.1356 。 -
目标化合物的制备方法参考化合物F1。
淡灰色固体(0.208 g,收率81%)。1H NMR(600 MHz, DMSO-d6)δ 11.90(s, 1H), 9.58(s, 1H), 9.40(s, 1H), 7.18(t, J=7.8 Hz, 1H), 7.07(d, J=7.8 Hz, 1H), 7.02(s, 1H), 6.89(d, J=7.8 Hz, 1H), 6.76(s, 1H), 6.24(s, 1H), 2.97-2.90(m, 1H), 2.19(s, 3H), 0.92(s, 3H), 0.91(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 157.41, 155.18, 154.64, 145.73, 138.66, 134.66, 129.03, 128.83, 127.82, 127.77, 125.93, 124.18, 105.02, 102.87, 25.91, 22.92(s, 2C), 21.20。
-
目标化合物的制备方法参考化合物F1。
淡灰色固体(0.202 g,收率78%)。1H NMR(600 MHz, DMSO-d6)δ 11.99(s, 1H), 9.64(s, 1H), 9.55(s, 1H), 7.32-7.26(m, 2H), 7.22-7.18(m, 1H), 7.12(d, J=7.8 Hz, 1H), 6.69(s, 1H), 6.28(s, 1H), 2.98-2.90(m, 1H), 2.11(s, 3H), 0.90(d, J=6.9 Hz, 3H), 0.89(d, J=6.9 Hz, 3H). 13C NMR(151 MHz, DMSO-d6)δ 157.34, 155.37, 154.37, 145.95, 136.71, 133.65, 131.07, 129.33, 129.31, 126.98, 126.93, 125.88, 104.61, 102.87, 25.73, 22.91, 22.83, 17.72。
-
目标化合物的制备方法参考化合物F1。
淡灰色固体(0.183 g,收率71%)。1H NMR(600 MHz, DMSO-d6)δ 11.93(s, 1H), 9.59(s, 1H), 9.35(s, 1H), 7.27-7.17(m, 4H), 6.88(s, 1H), 6.25(s, 1H), 3.06-2.98(m, 1H), 1.03(s, 3H), 1.02(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 161.44(d, J=244.6 Hz), 157.52, 154.88, 154.54, 145.69, 131.14, 129.04(d, J=8.6 Hz, 2C), 128.07, 125.96, 115.96(d, J=22.8 Hz, 2C), 105.11, 102.74, 25.99, 22.96(s, 2C). HRMS(ESI) m/z: calcd for C17H15FN3O3 [M-H]−
328.1103 , Found328.1107 。 -
目标化合物的制备方法参考化合物F1。
淡灰色固体(0.205 g,收率79%)。1H NMR(600 MHz, DMSO-d6)δ 12.00(s, 1H), 9.64(s, 1H), 9.39(s, 1H), 7.42-7.37(m, 1H), 7.19-7.12(m, 1H), 7.09-7.05(m, 1H), 7.00-6.97(m, 1H), 6.91(s, 1H), 6.26(s, 1H), 3.08-2.97(m, 1H), 1.03(s, 3H), 1.02(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 162.08(d, J=243.7 Hz), 157.63, 154.89, 154.32, 145.48, 136.33(d, J=10.6 Hz), 130.66(d, J=9.0 Hz), 128.15, 126.12, 122.80, 114.81(d, J=20.8 Hz), 114.02(d, J=24.0 Hz), 105.12, 102.78, 26.04, 22.98(s, 2C)。
-
目标化合物的制备方法参考化合物F1。
淡灰色固体(0.196 g,收率76%)。1H NMR(600 MHz, DMSO-d6)δ 12.00(s, 1H), 9.63(s, 1H), 9.41(s, 1H), 7.44-7.39(m, 1H), 7.33-7.28(m, 1H), 7.28-7.24(m, 1H), 7.22-7.16(m, 1H), 6.84(s, 1H), 6.25(s, 1H), 3.02-2.94(m, 1H), 0.99(s, 3H), 0.98(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 157.84(d, J=250.5 Hz), 157.58, 155.03, 154.36, 146.04, 131.09(d, J=7.6 Hz), 130.60, 127.52, 125.94, 125.04, 122.42(d, J=12.6 Hz), 116.63(d, J=19.4 Hz), 104.68, 102.74, 25.93, 22.93(s, 2C)。
-
目标化合物的制备方法参考化合物F1。
淡灰色固体(0.211 g,收率80%)。1H NMR(600 MHz, DMSO-d6)δ 11.97(s, 1H), 9.61(s, 1H), 9.38(s, 1H), 7.47-7.37(m, 1H), 7.35-7.27(m, 1H), 7.15-7.09(m, 1H), 6.89(s, 1H), 6.24(s, 1H), 3.08-2.95(m, 1H), 1.04(s, 3H), 1.03(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 161.52(dd, J=247.8, 11.8 Hz), 157.35(dd, J=252.3, 13.5 Hz), 156.92, 154.09, 153.52, 145.32, 130.92(d, J=10.1 Hz), 127.03, 125.21, 118.44(d, J=12.3 Hz), 111.29(d, J=21.8 Hz), 104.41(t, J=25.5 Hz), 103.92, 101.91, 25.23, 22.20(s, 2C). HRMS(ESI)m/z: calcd for C17H14F2N3O3 [M-H]−
346.1009 , Found346.1002 。 -
目标化合物的制备方法参考化合物F1。
淡灰色固体(0.219 g,收率81%)。1H NMR(600 MHz, DMSO-d6)δ 12.03(s, 1H), 9.63(s, 1H), 9.32(s, 1H), 7.73(d, J=8.5 Hz, 2H), 7.36(d, J=8.5 Hz, 2H), 6.93(s, 1H), 6.23(s, 1H), 3.05-2.96(m, 1H), 1.03(s, 3H), 1.02(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 157.79, 154.65, 154.23, 145.39, 138.56, 128.20, 128.02(q, J=32.0 Hz, 2C), 126.95(s, 2C), 126.25, 126.23, 124.44(q, J=272.0 Hz), 105.10, 102.77, 26.09, 22.93(s, 2C). HRMS(ESI)m/z: calcd for C18H15F3N3O3 [M-H]−
378.1071 , Found 378.1076. -
目标化合物的制备方法参考化合物F1。
淡灰色固体(0.203 g,收率75%)。1H NMR(600 MHz, DMSO-d6)δ 12.05(s, 1H), 9.63(s, 1H), 9.36(s, 1H), 7.66(d, J=7.9 Hz, 1H), 7.60(t, J=7.9 Hz, 1H), 7.52(s, 1H), 7.46(d, J=7.9 Hz, 1H), 6.95(s, 1H), 6.25(d, J=2.4 Hz, 1H), 3.07-2.99(m, 1H), 1.04(s, 3H), 1.02(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 157.72, 154.74, 154.30, 145.43, 135.54, 130.38, 129.84(q, J=32.2 Hz), 128.25, 126.30, 124.46, 124.12(q, J=272.4 Hz), 123.25, 123.22, 105.02, 102.72, 26.04, 22.94(s, 2C)。
-
目标化合物的制备方法参考化合物F1。
淡灰色固体(0.211 g,收率78%)。1H NMR(600 MHz, DMSO-d6)δ 12.01(s, 1H), 9.67(s, 1H), 9.61(s, 1H), 7.83(d, J=7.7 Hz, 1H), 7.75(t, J=7.7 Hz, 1H), 7.66(t, J=7.7 Hz, 1H), 7.54(d, J=7.7 Hz, 1H), 6.58(s, 1H), 6.25(s, 1H), 2.95-2.83(m, 1H), 0.85(d, J=6.9 Hz, 3H), 0.82(d, J=6.9 Hz, 3H). 13C NMR(151 MHz, DMSO-d6)δ 157.46, 155.48, 154.71, 145.83, 133.99, 132.66, 132.03, 130.67, 127.97, 127.92(q, J=30.5 Hz), 126.91, 125.81, 123.52(q, J=273.9 Hz), 104.03, 102.83, 25.60, 22.84(s, 2C)。
-
以化合物29o为原料制备中间体31o。1H NMR(600 MHz, CDCl3)δ 8.38(s, 1H), 7.94(s, 1H), 7.35(d, J=8.9 Hz, 2H), 6.89(d, J=8.9 Hz, 2H), 6.75(s, 1H), 3.81(bs, 2H), 1.48(s, 9H)。
取中间体31o(0.642 g, 2.41 mmol, 1.0 equiv)溶解于无水乙醇/1,4-二氧六环(1/1, V/V, 50 ml)中,在回流条件下搅拌反应2 h,后依次加入NaOH(0.58 g, 14.5 mmol, 6.0 equiv)和K3Fe(CN)6(2.38 g, 7.23 mmol, 3.0 equiv),继续在回流条件下搅拌反应12 h,向反应液中加入NaOH(0.193 g, 4.82 mmol, 2.0 equiv)和H2O(20 ml),并继续在回流条件下搅拌反应12 h,待反应液降至室温后,过滤,滤液加入150 ml的水稀释,以1 mol/L HCl水溶液调节pH至7左右,加入乙酸乙酯(200 ml×2)萃取,收集乙酸乙酯层,无水Na2SO4干燥,过滤,滤液经减压除去溶剂,得残余物以流动相(CH2Cl2/CH3OH, 100/0-100/8, V/V)在硅胶柱层析中纯化得棕色固体状中间体32o(0.425 g, 收率35%)。1H NMR(600 MHz, DMSO-d6)δ 11.79(s, 1H), 7.40-7.30(m, 8H), 7.22(d, J=7.3 Hz, 2H), 7.11(s, 1H), 6.72(s, 1H), 6.67(d, J=8.6 Hz, 2H), 6.42(d, J=8.6 Hz, 2H), 5.21(s, 2H), 5.06(s, 2H), 4.94(s, 2H), 3.17-3.09(m, 1H), 1.08(s, 3H), 1.07(s, 3H)。
取中间体32o(0.40 g, 0.790 mmol, 1.0 equiv)溶解于无水甲醇(20 ml)中,向其中加入10%的Pd/C(0.042 g, 39.5μmol, 0.05 equiv),在H2条件下室温搅拌反应12 h,过滤,滤液经减压除去溶剂,得残余物以流动相(CH2Cl2/CH3OH, 100/1-100/5, V/V)在硅胶柱层析中纯化得淡黄色固体即为目标化合物F15(0.202 g, 收率78%)。1H NMR(600 MHz, DMSO-d6)δ 11.79(s, 1H), 9.62(s, 1H), 9.52(s, 1H), 6.80(d, J=8.6 Hz, 2H), 6.73(s, 1H), 6.50(d, J=8.6 Hz, 2H), 6.28(s, 1H), 5.26(bs, 2H), 2.98-2.87(m, 1H), 0.92(s, 3H), 0.91(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 157.19, 155.42, 155.03, 149.02, 146.25, 128.44(s, 2C), 127.35, 125.68, 122.56, 114.03(s, 2C), 104.89, 102.87, 25.84, 22.93(s, 2C). HRMS(ESI)m/z: calcd for C17H17N4O3 [M-H]−
325.1306 , Found325.1314 。 -
目标化合物的制备方法参考化合物F15。
淡黄色固体(0.220 g, 收率85%)。1H NMR(600 MHz, DMSO-d6)δ 11.87(s, 1H), 9.60(s, 1H), 9.50(s, 1H), 6.99(t, J=7.9 Hz, 1H), 6.79(s, 1H), 6.56-6.51(m, 1H), 6.47(t, J=2.1 Hz, 1H), 6.29(s, 1H), 6.26-6.24(m, 1H), 5.27(bs, 2H), 3.02-2.93(m, 1H), 0.96(s, 3H), 0.95(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 157.22, 155.38, 154.54, 149.78, 145.69, 135.32, 129.48, 127.28, 125.73, 114.57, 113.85, 112.94, 104.78, 102.86, 25.84, 22.91(s, 2C)。
-
目标化合物的制备方法参考化合物F15。
淡黄色固体(0.183 g, 收率71%)。1H NMR(600 MHz, DMSO-d6)δ 11.92(s, 1H), 9.69(s, 1H), 9.65(s, 1H), 7.11-7.06(m, 1H), 6.87-6.83(m, 1H), 6.80-6.76(m, 1H), 6.74(s, 1H), 6.55-6.50(m, 1H), 6.29(s, 1H), 3.62(bs, 2H), 2.92-2.83(m, 1H), 0.83(d, J=6.9 Hz, 3H), 0.76(d, J=6.9 Hz, 3H). 13C NMR(151 MHz, DMSO-d6)δ 157.11, 155.64, 154.50, 145.87, 130.19, 129.96, 126.10, 125.87, 119.24, 116.71, 116.37, 104.02, 102.90, 25.58, 22.86, 22.82。
-
目标化合物的制备方法参考化合物F15。
淡灰色固体(0.196 g, 收率74%)。1H NMR(600 MHz, DMSO-d6)δ 13.02(bs, 1H), 12.00(s, 1H), 9.61(s, 1H), 9.30(s, 1H), 7.91(d, J=8.3 Hz, 2H), 7.28(d, J=8.3 Hz, 2H), 6.95(s, 1H), 6.24(s, 1H), 3.07–3.01(m, 1H), 1.06(s, 3H), 1.05(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 167.34, 157.68, 154.72, 154.25, 145.48, 138.52, 130.45, 130.07(s, 2C), 128.15, 126.26(s, 2C), 126.10, 105.23, 102.72, 26.07, 22.96(s, 2C). HRMS(ESI)m/z: calcd for C18H18N3O5 [M+H]+
356.1241 , Found356.1253 。 -
目标化合物的制备方法参考化合物F15。
淡灰色固体(0.220 g, 收率83%)。1H NMR(600 MHz, DMSO-d6)δ 13.14(bs, 1H), 11.99(s, 1H), 9.61(s, 1H), 9.34(s, 1H), 7.92-7.86(m, 1H), 7.79(t, J=1.8 Hz, 1H), 7.50(t, J=7.9 Hz, 1H), 7.41-7.33(m, 1H), 6.94(s, 1H), 6.25(s, 1H), 3.10-2.97(m, 1H), 1.05(s, 3H), 1.04(s, 3H). 13C NMR(151 MHz, DMSO-d6)δ 166.98, 157.59, 154.77, 154.45, 145.56, 135.08, 131.97, 130.80, 129.39, 128.59, 128.15, 127.67, 126.10, 105.16, 102.77, 26.05, 22.97(s, 2C)。
-
化合物体外协同抗耐药真菌活性测试采用美国临床和实验室标准协会推荐的棋盘式微量液基稀释法,实验步骤如下所示。
收集指数生长末期的真菌细胞,以真菌用RPMI
1640 培养基稀释至1×103 CFU/ml。按照每孔100 μl接种真菌稀释液至透明96孔板中,其中1~11列加入系列稀释的化合物,A-H行加入系列稀释的FLC(第12列作为阳性对照,孔中仅加入真菌稀释液不加入药物,第G行作为阴性对照,孔中不加入药物也以RPMI1640 培养基代替真菌稀释液,11列仅加入化合物不加入FLC,H行仅加入FLC不加入化合物),而后在35 ℃条件下孵育48 h。测定每孔在630 nm处的吸光度A,依据公式:抑制率%=(A阳性对照孔−A化合物孔)/(A阳性对照孔−A阴性对照孔)× 100%,计算各孔对应的抑制率。在96孔板中A1-F10区域内,如果某一孔对应的所有左上方孔和该孔的抑制率均大于80%,则该孔对应的化合物和FLC浓度分别作为FIC化合物和FICFLC,利用公式:FICI=(FIC化合物/MIC80化合物)+(FICFLC/MIC80FLC)(MIC80>64 μg/ml的化合物,MIC80以64 μg/ml计算),计算各化合物对应的FICI。
-
体外Hsp90α抑制活性测试采用荧光偏振法,实验步骤如下所示。
向黑色96孔板的每孔中加入Hsp90α(3.4 μg/ml)、FITC-Geldanamycin和系列稀释的化合物的实验用Buffer溶液100 μl(阳性对照孔不加入化合物,阴性对照孔既不加入Hsp90α也不加入化合物),而后在室温条件下孵育3 h。测定每孔在激发波长485 nm和发射波长530 nm下的荧光强度,根据荧光强度转换得荧光偏振值Fb。利用公式:抑制率=(Fb阳性对照孔−Fb化合物孔)/(Fb阳性对照孔−Fb阴性对照孔)× 100%,计算各浓度下化合物的抑制率,并以Graphpad软件计算化合物IC50值。
-
抗HEL和HL60的细胞活性测试步骤如下所示。
在透明96孔板中每孔加入50 μl接种HEL细胞(含10 000个)培养基悬液或HL60细胞(含20 000个)培养基悬液,在37 ℃和5% CO2的条件下培养24 h。向每孔加入含有系列浓度的化合物培养基溶液50 μl,继续在37 ℃和5% CO2的条件下培养48 h(阳性对照孔接种细胞不加入化合物,阴性对照孔不接种细胞也不加入化合物,并均以纯培养基代替)。向每孔中加入含有10 μl的CCK-8,在37 ℃和5% CO2的条件下培养30 min。测定每孔在450 nm处的吸光度A,依据公式:抑制率% =(A阳性对照孔−A化合物孔)/(A阳性对照孔−A阴性对照孔)× 100%,计算各浓度下化合物的抑制率,并以Graphpad软件计算化合物的IC50值。
抗A549的细胞活性测试步骤如下所示。
按照每孔5 000个细胞数接种A549的细胞培养基悬液100 μl于透明96孔板中,在37 ℃和5% CO2的条件下培养24 h。吸弃每孔培养基,向每孔加入含有不同浓度的化合物培养基溶液100 μl,在37 ℃和5% CO2的条件下继续培养48 h(阳性对照孔接种细胞不加入化合物,阴性对照孔不接种细胞也不加入化合物且以等体积纯培养基代替)。吸弃每孔培养基,向每孔中加入含有10% CCK-8的培养基溶液100 μl,在37 ℃和5% CO2的条件下继续培养30 min。测定每孔在450 nm处的吸光度A,依据公式:抑制率%=(A阳性对照孔−A化合物孔)/(A阳性对照孔−A阴性对照孔)× 100%,计算各浓度下化合物的抑制率,并以Graphpad软件计算化合物IC50。
-
收集指数生长末期的C. albicans 0304103,并以真菌用RPMI
1640 培养基稀释至1 × 104 CFU/ml。按照每管5 ml接种真菌稀释液至培养管中,加入不同浓度的化合物和32 μg/ml的FLC,后在30 ℃和200 r/min条件下的恒温振荡箱中培养。取0、3、6、9、12和24 h时各组真菌培养液100 μl,以PBS稀释至适宜浓度后,均匀涂布在含有SDA固体培养基的培养皿中,各组每个时间点均涂布3次,而后在30 ℃条件下培养48 h。对培养皿中生成的菌落数计数,并绘制时间-菌落数折线图。 -
挑取C. albicans 0304103单菌落接种于1 ml的YEPD培养基,在30 ℃的条件下培养16 h。吸取10 μl真菌培养液,分别接种于含有化合物,FLC和空白组的YEPD培养基(50 ml)中,然后在30 ℃条件下继续培养24 h。离心收集各组真菌细胞,参照柱式真菌RNAout试剂盒提取各组真菌总RNA,并测定各组提取的总RNA浓度。以提取的总RNA为模板参照反转录试剂盒制备各组cDNA。以获得的cDNA参照qPCR试剂盒,在40个循环内扩增各组基因以计算CT值。根据公式:相对表达量=2(−ΔΔCT),计算不同组间各基因的表达倍数。本实验中所使用的内参基因为ACT1(表1)。
表 1 Real time RT-PCR实验所用的基因引物序列
名称 序列 ERG11-F ACTCATGGGGTTGCCAATGT ERG11-R GAGCAGCATCACGTCTCCAA CDR1-F TCCACGGTCGTGAATTCCAATGTG CDR1-R GCCAGCAACAGGACCAGCTTC CDR2-F GCTACTGCCATGTCACTCTCCAC CDR2-R GGACAACTGTGCTTCCAGGAGTAG MDR1-F CCACTGGTGGTGCAAGTGTT MDR1-R TCGTTACCGGTGATGGCTCT ALS1-F GTGTCGGTTGTCAGAAGAGC ALS1-R TTGTTCACGTTGAGCCATGG ALS3-F ACTTTGTGGTCTACAACTTGGG ALS3-R CCAGATGGGGATTGTAAAGTGG HWP1-F CTGAACCTTCCCCAGTTGCT HWP1-R CGACAGCACTAGATTCCGGA EAP1-F TCCTACACGACTGACACTGC EAP1-R TGACACCCGTAGTTACTGCTG BCR1-F TCCTTTACGTGCACCACCTC BCR1-R ATGCCGACGATTCAGCTGAT ACE2-F ACTTTGTGGTCTACAACTTGGG ACE2-R CCAGATGGGGATTGTAAAGTGG RLM1-F GTGCCTGCGAATGTTCCAAA RLM1-R TGCATTGCTTCCTCCTGTCA ZAP1-F TACCGCGACTACAAACCACC ZAP1-R TGCCCCTGTTGCTCATGTTT ACT1-F GGTTTGGAAGCTGCTGGTAT ACT1-R ACCACCAATCCAGACAGAGT -
离心收集指数生长末期的C. albicans 0304103,以真菌用RPMI 1
640 培养基稀释至1×106 CFU/ml,后以每孔100 μl接种至透明96孔板中,在37 ℃下培养90 min(阴性对照组不接种真菌)。吸弃每孔培养基,以PBS清洗2次,向每孔中加入系列稀释的化合物RPMI1640 培养基溶液(阳性对照孔和阴性对照孔均加入不含化合物培养基)。后继续在37 ℃下培养24 h。吸弃每孔培养基,以PBS清洗2次,向每孔中加入甲萘醌(3 μmol/L)和XTT(0.5 mg/ml)的PBS溶液100 μl,继续在37 ℃下培养3 h。吸取各孔培养液80 μl至另一透明96孔板中,读取各孔在492 nm处的吸光度值A。根据吸光度计算各浓度化合物下的生物被膜形成率,形成率=(A化合物孔−A阴性对照孔)/(A阳性对照孔−A阴性对照孔)×100%。 -
离心收集指数生长末期的C. albicans 0304103,以Spider培养基稀释真菌至1×105 CFU/ml。吸取2 ml菌悬液接种在12孔板中,并加入不同浓度化合物,在37 ℃下培养3 h。使用无目镜倒置显微镜观察并记录各组真菌的细胞形态。
-
吸取1 ml含HEL细胞的培养基悬液(含3×105个细胞)接种于24孔板中,在37 ℃和5% CO2的条件下培养24 h。向每孔中加入含有系列浓度的化合物培养基溶液1 ml(对照孔不加入化合物),继续在37 ℃和5% CO2的条件下培养48 h。1 000 r/min离心5 min收集每孔细胞悬液,并以PBS清洗两次。向收集的细胞中加入300 μl Binding Buffer结合液以及10 μl PI染料,混合均匀,在室温下避光孵育15 min,以流式细胞仪检测各组细胞。
-
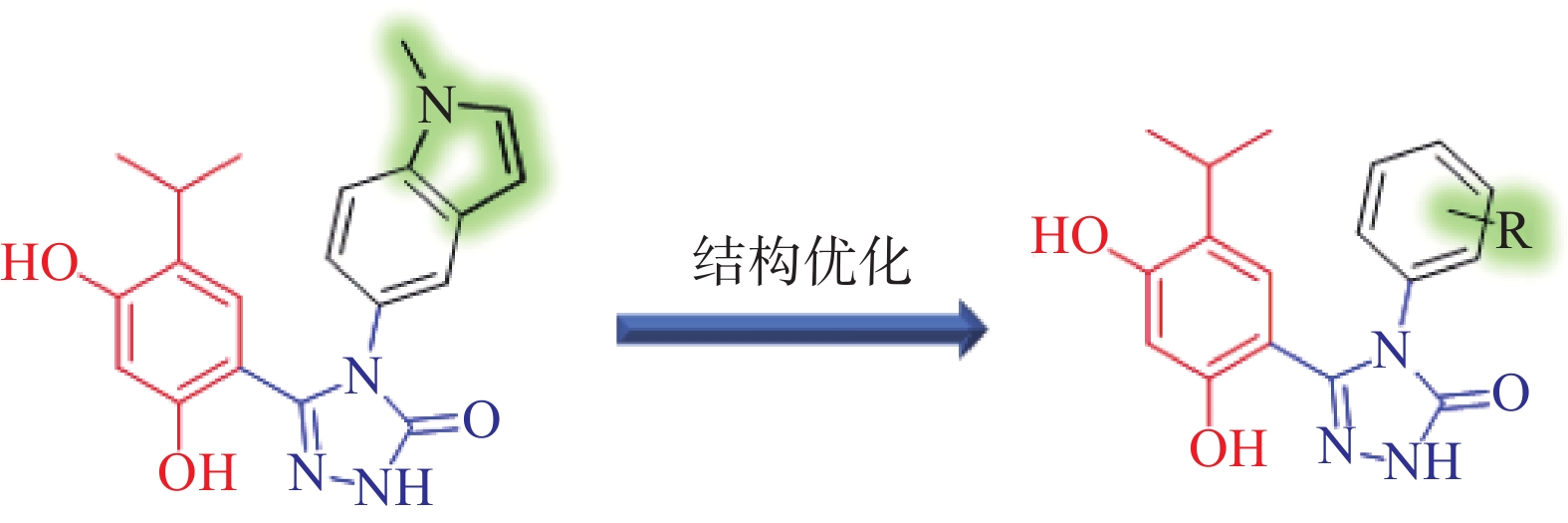
分析Ganetespib与人Hsp90α的复合物晶体结构(PDB:3UTH),可发现其分子中的间二酚部分和三唑酮部分与Hsp90α结合较为紧密(如间二酚部分的2’位酚羟基与Asp93,三唑酮的1位N原子与Thr184均存在氢键结合),而甲基吲哚片段中苯环一侧也可与Lys58存在阳离子-π作用,吡咯环一侧则处于溶剂暴露区[9]。近期Ganetespib的类似物AUY922同C. albicans Hsp90复合物晶体结构(PDB:6CJS)也已见报道[10]。该晶体结构显示,AUY922同C. albicans Hsp90的结合模式与Ganetespib同人Hsp90α之间的结合相似,结构中AUY922的间二酚部分的2’位酚羟基和噁(排版造字:口字旁的恶)唑环中1位O原子可分别与Asp82和Thr174形成氢键作用,而另一侧连接有吗啉的苯环与Lys47具有阳离子-π作用。因此,本研究拟计划保留Ganetespib重要的结合部分(间二酚部分、三唑酮部分与吲哚的苯环一侧),将吡咯环替换为新的基团(即吲哚环替换为含不同取代基的苯环)以设计新型Hsp90抑制剂,以期发现活性更优的抗真菌与抗肿瘤双重作用化合物(图2)。
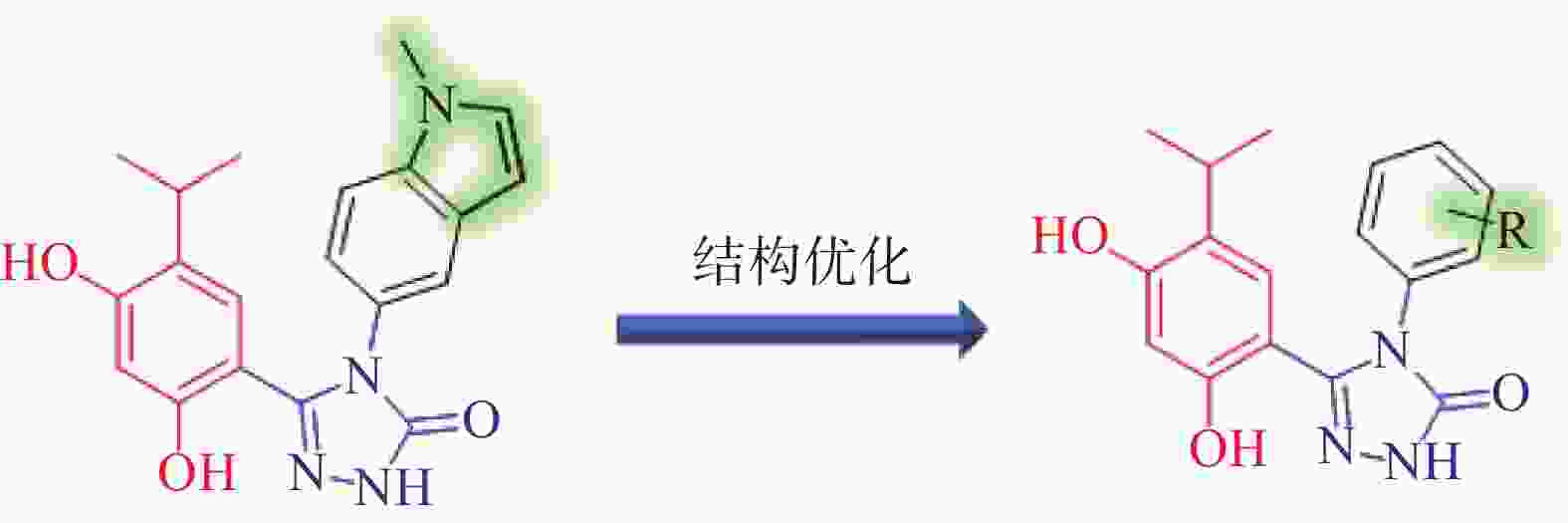
图 2 新型Hsp90抑制剂的设计
-
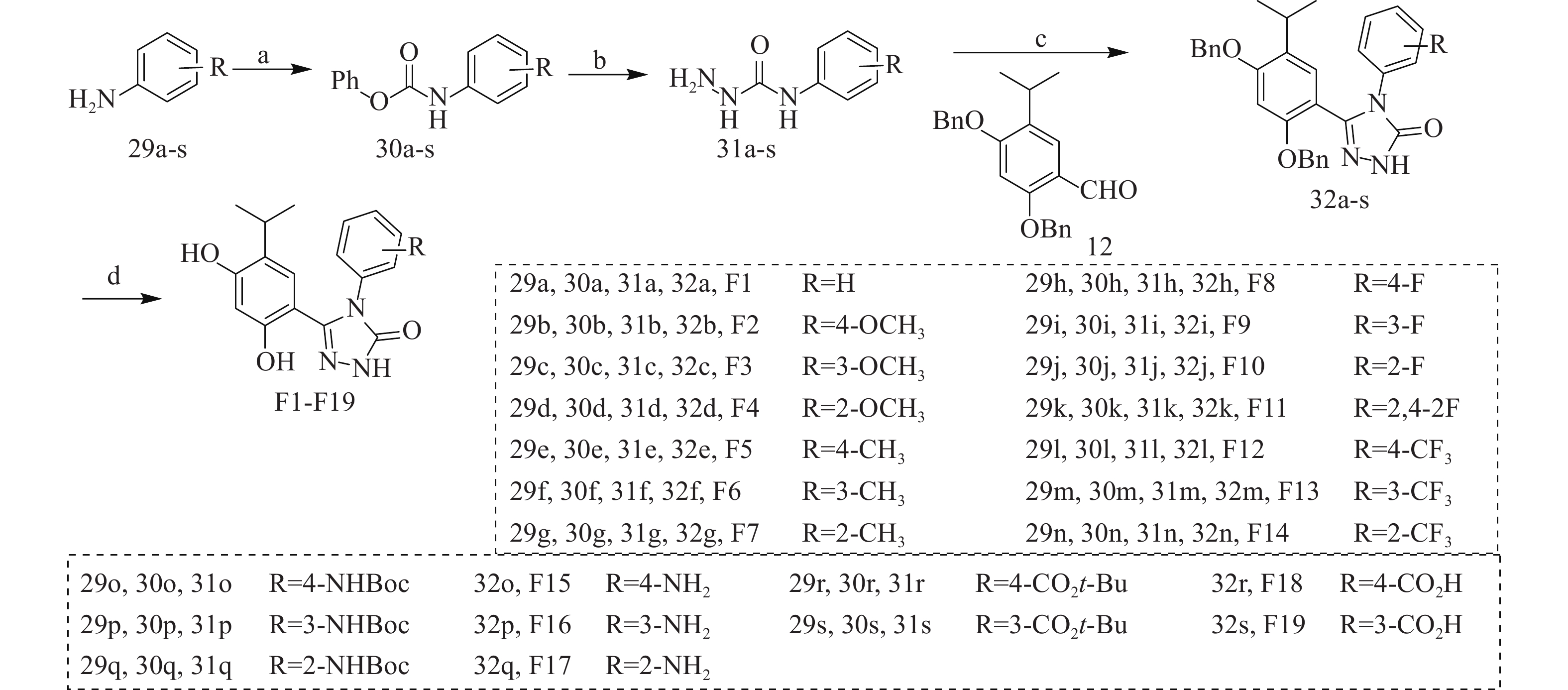
以含不同取代基的苯胺(29a-s)为原料,在三乙胺作用下与氯甲酸苯酯反应得到中间体30a-s,在回流条件下与水合肼反生肼解反应得到中间体31a-s,再经三步反应得到中间体32a-s,最后经H2作用下去除苄基保护基得到目标化合物F1-F19(图3)。
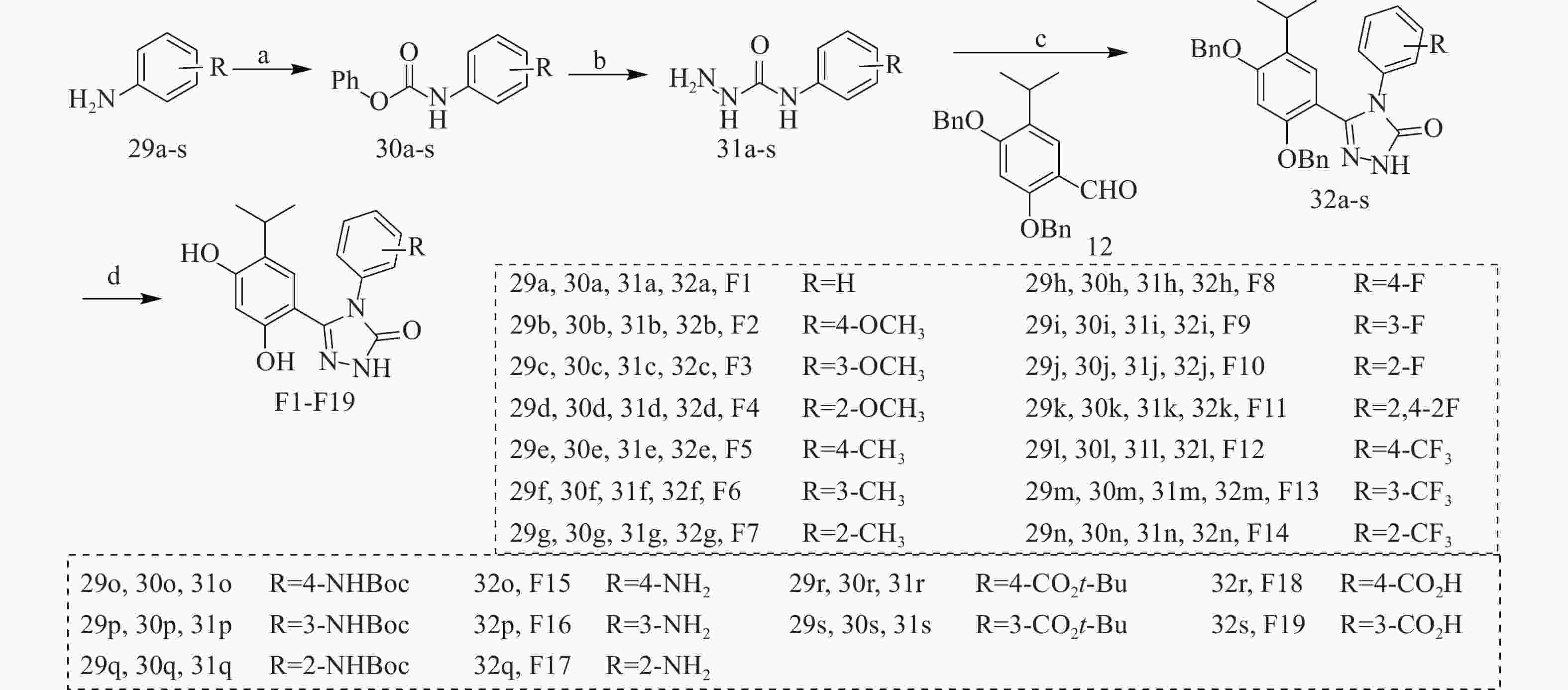
图 3 目标化合物的合成路线
-
目标化合物的体外活性评价包括对人源Hsp90α的抑制活性评价、通过棋盘式微量液基稀释法测试的化合物联合FLC的体外协同抗耐药真菌活性以及抗肿瘤细胞活性评价。其中,耐药真菌选择临床分离的耐药C. albicans(菌编号:0304103);肿瘤细胞选择2株血液肿瘤细胞系HEL(人红白细胞白血病细胞)和HL60(人早幼粒白血病细胞),1株人肺癌细胞系(A549)。实验中以先导化合物Ganetespib作为对照药物。
通过将Ganetespib的分子中甲基吲哚结构替换为含有不同取代基的苯环得到了目标化合物,该类化合物的体外活性如表2所示。在1 μmol/L的浓度下,目标化合物对Hsp90α均具有强的抑制作用(抑制率均≥94%)。
表 2 化合物F1-F19的Hsp90α抑制活性、体外协同抗耐药真菌及抗肿瘤活性
化合物 取代基(R) Hsp90α C. albicans
0304103(μg/ml)HEL HL60 A549 抑制率(%,1 μmol/L) MIC80 FICI IC50(μmol/L) IC50(μmol/L) IC50(μmol/L) F1 H 99 >64 0.047 0.049±0.003 0.13±0.05 0.15±0.024 F2 4-OCH3 99 >64 0.047 0.026±0.003 0.17±0.014 0.32±0.031 F3 3-OCH3 97 >64 0.047 0.025±0.002 0.061±0.011 0.15±0.022 F4 2-OCH3 97 >64 0.063 0.048±0.007 0.069±0.005 0.55±0.15 F5 4-CH3 97 >64 0.047 0.021±0.002 0.067±0.005 0.23±0.026 F6 3-CH3 97 >64 0.047 0.052±0.002 0.12±0.017 0.22±0.02 F7 2-CH3 98 >64 0.023 0.107±0.019 0.28±0.029 0.79±0.076 F8 4-F 95 >64 0.125 0.138±0.012 0.325±0.026 0.26±0.008 F9 3-F 97 >64 0.063 0.115±0.012 0.25±0.008 0.25±0.063 F10 2-F 95 >64 0.047 0.037±0.002 0.083±0.004 0.40±0.067 F11 2,4-2F 94 >64 0.188 0.153±0.012 0.466±0.012 0.76±0.012 F12 4-CF3 96 >64 2 0.033±0.001 0.115±0.007 0.10±0.022 F13 3-CF3 97 >64 0.094 0.085±0.009 0.361±0.024 0.47±0.1 F14 2-CF3 98 >64 − 0.408±0.022 0.650±0.014 0.22±0.02 F15 4-NH2 99 >64 0.063 0.047±0.003 0.055±0.003 0.65±0.14 F16 3-NH2 97 >64 0.125 0.070±0.034 0.077±0.006 0.25±0.063 F17 2-NH2 96 >64 0.141 0.120±0.025 0.135±0.013 0.63±0.068 F18 4-CO2H 94 >64 − 5.5±0.73 16±2.9 43±3 F19 3-CO2H 95 >64 − 16±1.2 13±0.47 21±2.1 Ganetespib − 99 >64 0.023 0.021±0.002 0.023±0.002 0.11±0.019 体外协同抗耐药真菌活性显示,绝大部分目标化合物都具有与FLC的协同作用(FICI<0.5)。整体上看,苯环上取代基的电子效应对协同抗耐药真菌活性的影响较大,如苯环上无取代基(F1)或含有供电子基团如甲氧基(F2-F4)、甲基(F5-F7)、氨基(F15-F17)或弱吸电子基团如氟取代(F8-F10)的化合物要比含有强吸电子基团如三氟甲基(F12-F14)和羧基(F18-F19)的化合物协同抗耐药真菌活性强,后者大部分化合物(F12, F14, F18-F19)甚至丧失与FLC的协同作用(FICI > 0.5);此外,含有单个吸电子基团的单氟取代的化合物(F8-F10)也要强于含有两个氟取代的化合物(F11)。对比同一取代基在苯环上的取代位置,可发现取代位点的不同对化合物的协同抗耐药真菌活性影响不大。
目标化合物的体外抗肿瘤活性显示,除含有强吸电子效应和负电荷的羧基取代化合物(F18-F19, IC50=5.5~43 μmol/L)外均呈现出强的抗肿瘤活性(IC50=0.021~0.8 μmol/L)。相比于协同抗耐药真菌活性,目标化合物分子中苯环上取代基的电子效应对抗肿瘤活性影响不大,如含强吸电子基团三氟甲基的化合物F12-F14(IC50=0.03~0.8 μmol/L)对同一肿瘤细胞系的活性仅稍弱于含供电子基团甲氧基的化合物F2-F4(IC50=0.02~0.6 μmol/L)。但苯环上取代基位置对活性影响较大,若苯环的2位上含有较大体积的取代基如甲氧基(F4)、甲基(F7)、三氟甲基(F14)、氨基(F17),其抗肿瘤活性要明显弱于这些取代基在苯环3位和4位取代的化合物,然而较小空间体积的氟取代化合物(F8、F9与F10比较)则几乎不受取代位点的影响。
由于化合物F3和F5的体外协同抗耐药真菌活性与抗肿瘤活性均较强,因此选择这两个化合物开展进一步的抗耐药真菌与肿瘤细胞的作用机制研究。
-
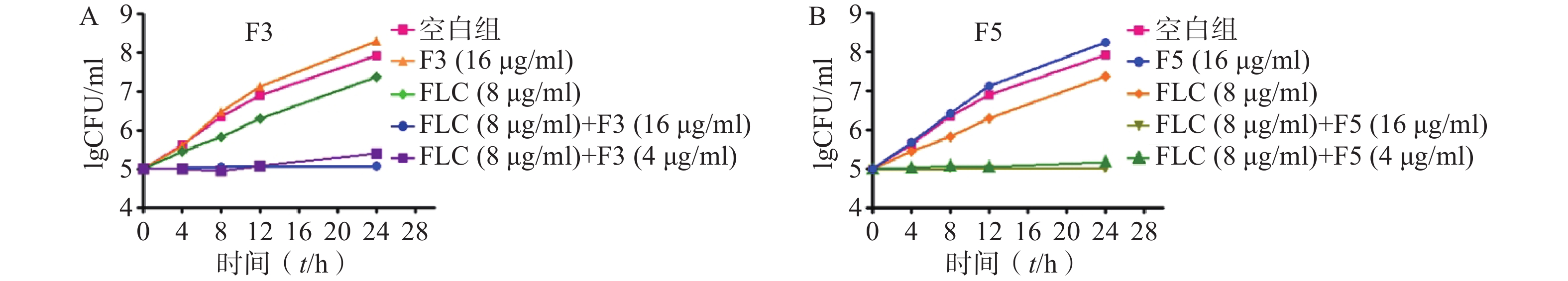
为进一步验证化合物F3和F5抗真菌作用机制,开展了时间-杀菌曲线实验。结果显示,在化合物F3(16 μg/ml)、F5(16 μg/ml)以及FLC(8 μg/ml,CLSI推荐的敏感性折点浓度)作用下,真菌能够正常生长,与化合物无直接抗真菌活性结果一致。而化合物F3和F5(4 μg/ml)与FLC(8 μg/ml)联合作用下,真菌生长几乎完全抑制(图4)。上述结果表明,化合物F3和F5能够恢复耐药真菌对FLC的敏感性,与设计思想吻合。
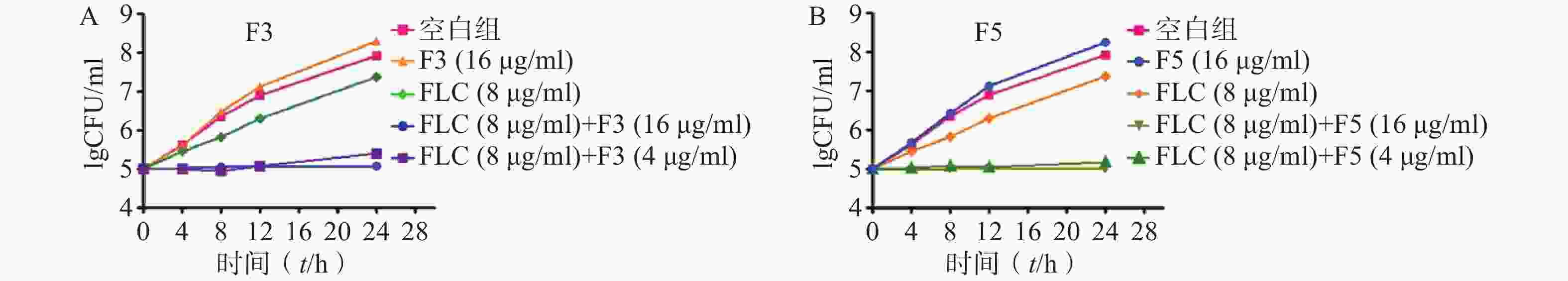
图 4 时间杀菌曲线
-
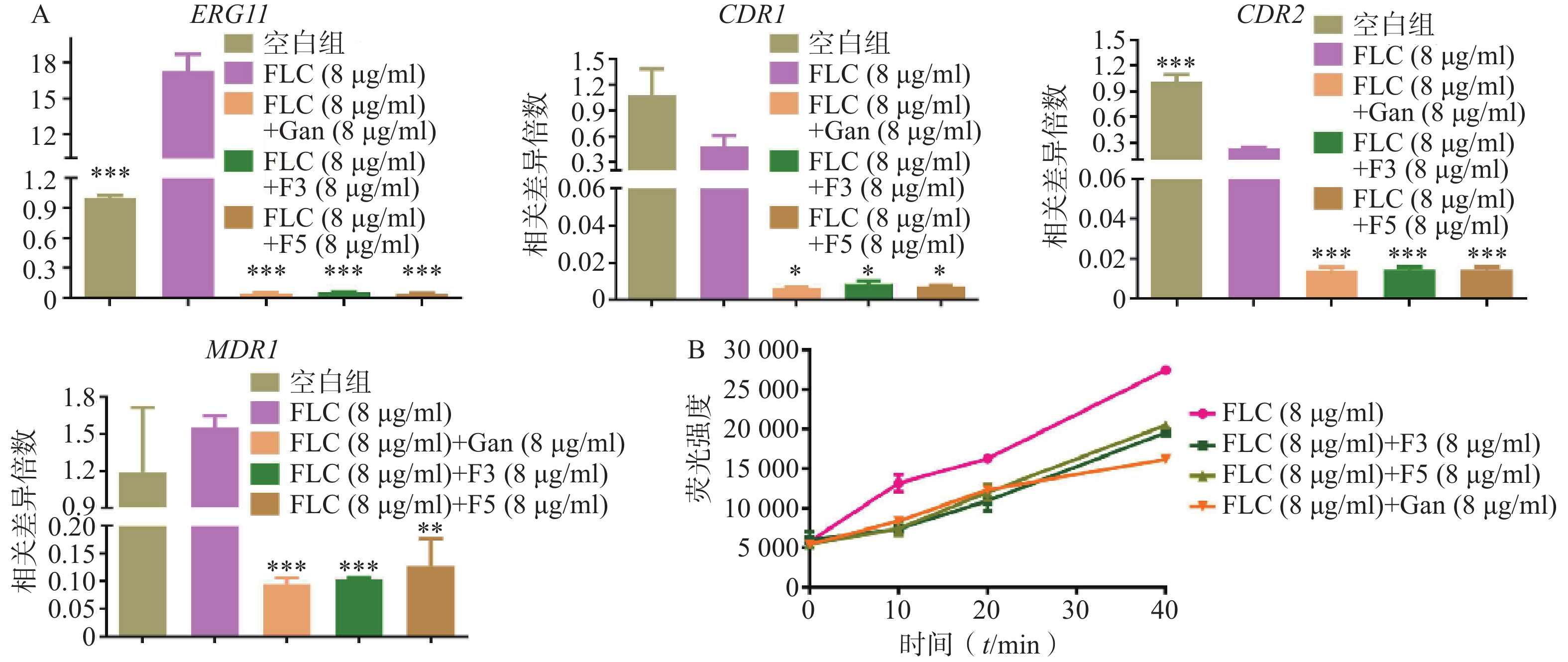
利用real time RT-PCR技术,对化合物F3(8 μg/ml)、F5(8 μg/ml)和先导化合物Ganetespib协同FLC(8 μg/ml)作用下白念珠菌的耐药相关基因如CYP51基因(ERG11)和外排泵相关基因(CDR1、CDR2、MDR1)的表达量进行了检测。结果如图5A所示:相比于FLC的单独用药组,化合物F3、F5以及先导化合物Ganetespib与FLC的联合用药组的ERG11、CDR1、CDR2和MDR1基因表达水平均显著下调。
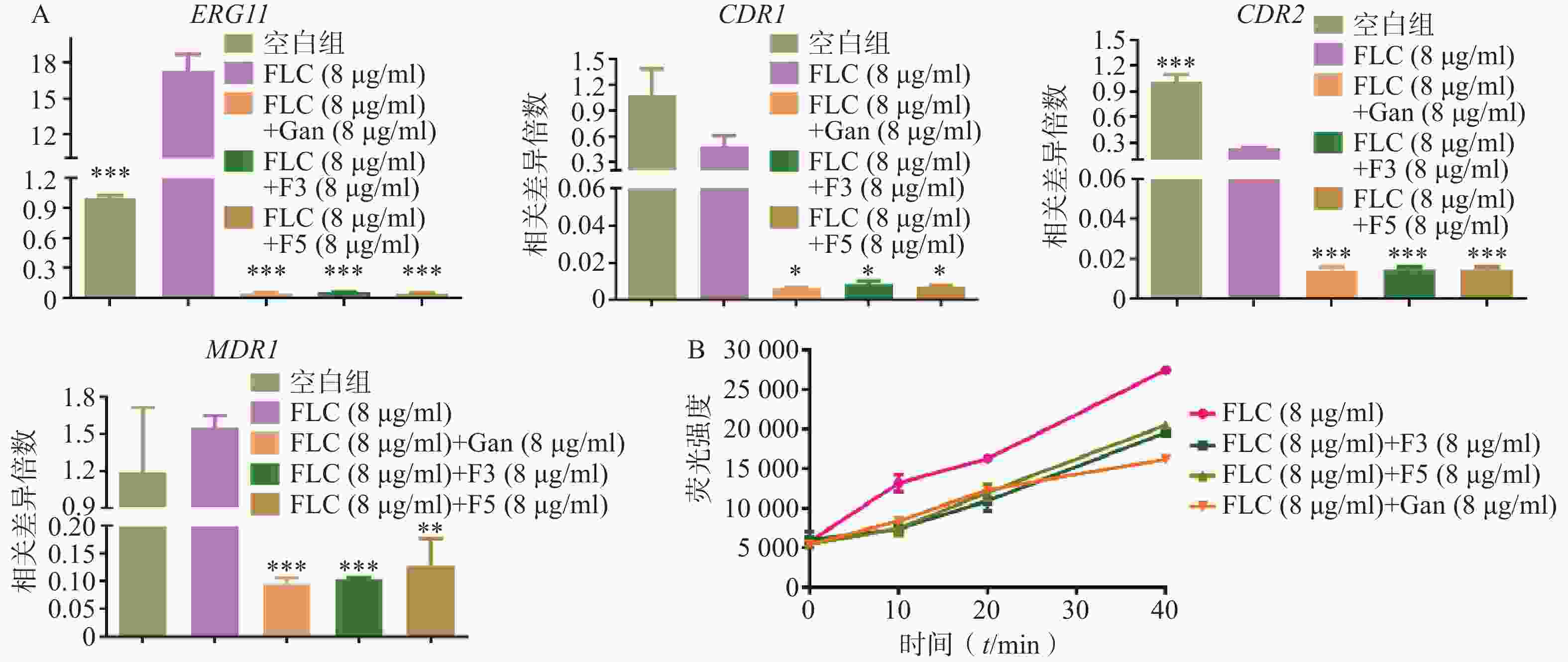
图 5 化合物对真菌外排泵的抑制
为验证化合物对药物外排泵基因表达的影响,进一步开展了Rh6G外排试验(图5B)。结果显示:化合物F3、F5和Ganetespib协同FLC组在多个时间点的Rh6G外排量均明显低于FLC组,这与它们在联合作用下真菌外排泵基因表达明显低于FLC组的real time RT-PCR实验结果相一致。
-
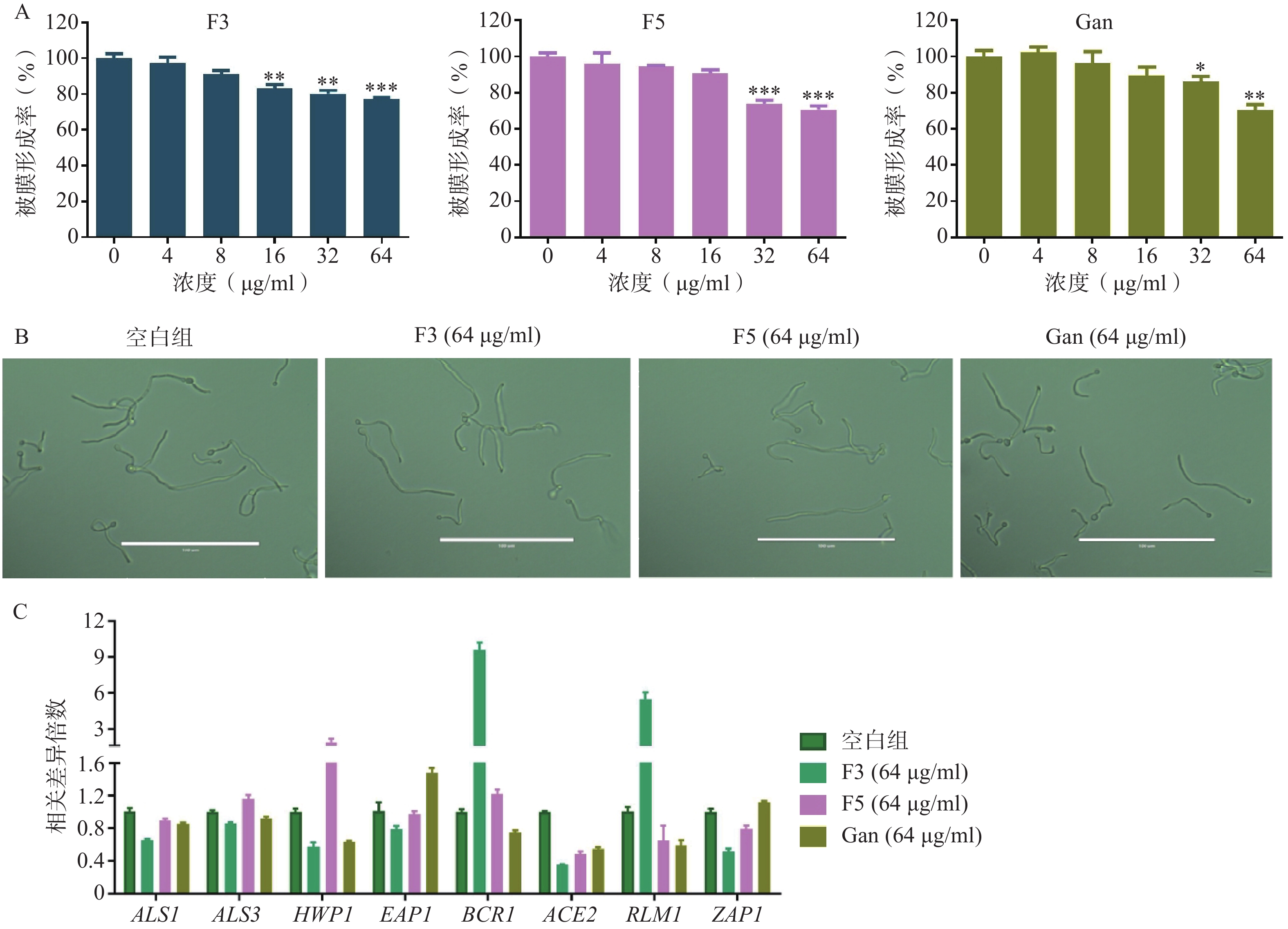
真菌生物被膜的形成既是真菌产生耐药的重要因素,也是重要的毒力因子。通过XTT还原法,对化合物F3、F5以及先导化合物Ganetespib作用下耐药白念珠菌生物被膜的形成率进行了测定。结果表明,化合物F3、F5以及Ganetespib在高浓度下对生物被膜的形成均具有抑制作用,如在64 μg/ml的浓度下,生物被膜形成率分别为77.3%、70.8%和70.4%(图6A)。
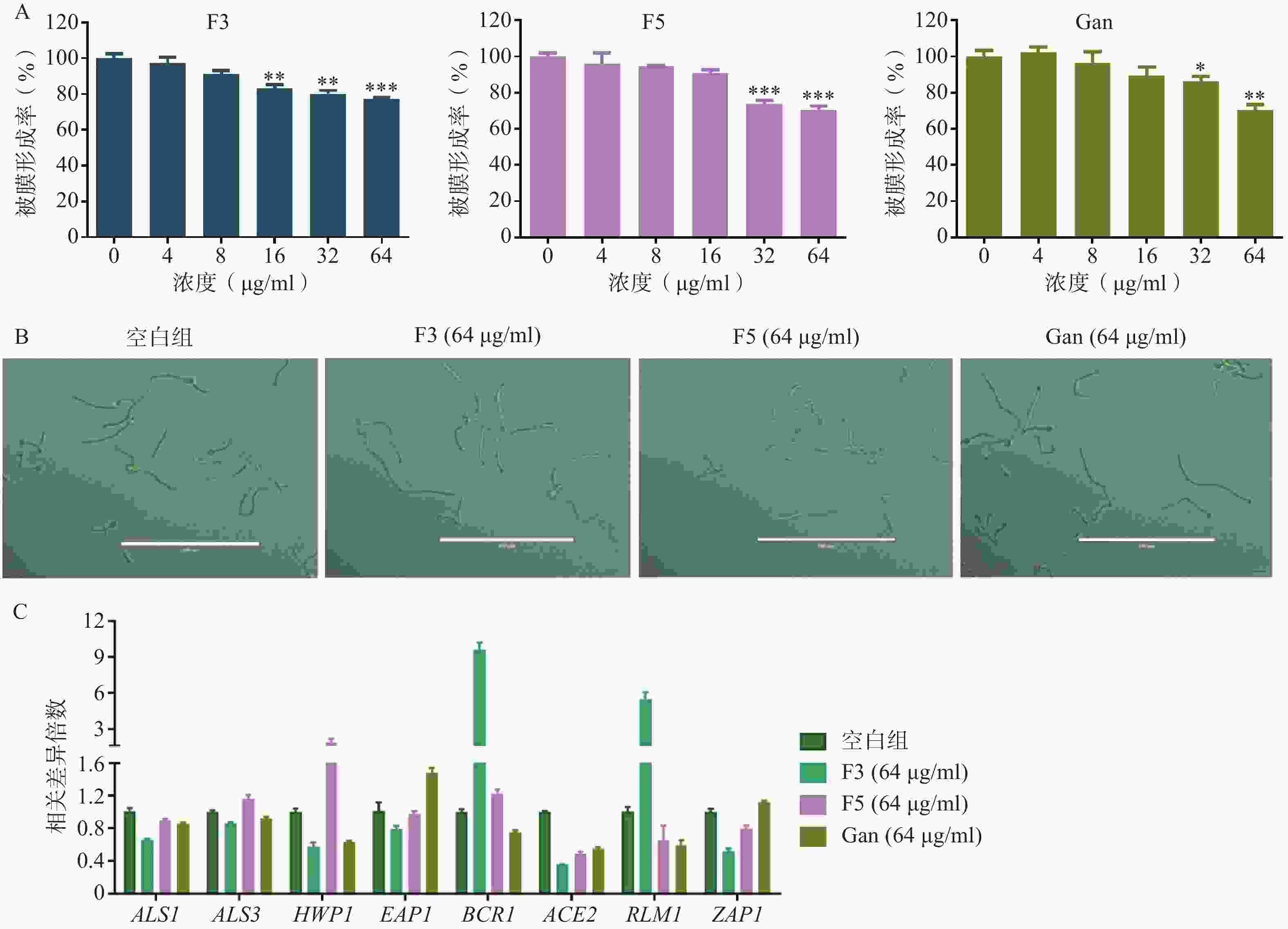
图 6 化合物对真菌生物被膜和菌丝的抑制
菌丝态真菌的产生是影响生物被膜形成的重要因素。为研究化合物F3、F5以及先导化合物Ganetespib抑制生物被膜形成的作用机制,通过倒置显微镜观察了化合物F3、F5以及先导化合物Ganetespib在64 μg/ml的浓度下,耐药真菌的形态上变化(图6B)。结果显示,与空白组相比,这些化合物作用后的真菌形态均无明显区别。提示化合物F3、F5以及先导化合物Ganetespib可能不通过抑制菌丝态真菌的产生而抑制生物被膜的形成。
真菌黏附作用的形成与胞外聚合物的产生也是影响生物被膜形成的重要因素。为进一步地探究化合物抑制生物被膜形成的作用机理,利用real time RT-PCR技术对这两种生物被膜形成因素的相关基因进行了考察(图6C)。基因ALS1、ALS3、HWP1、EAP1、BCR1和ACE2均编码影响真菌形成黏附作用相关的蛋白从而影响生物被膜的形成,基因RLM1和ZAP1则编码调节胞外聚合物产生的转录因子进而影响生物被膜的形成[11-18]。针对这些基因的real time RT-PCR实验结果表明,相比于空白组,化合物F3可显著引起ALS1、ALS3、HWP1、ACE2、ZAP1下调和BCR1、RLM1上调,化合物F5则显著引起ACE2、ZAP1下调和HWP1上调,而先导化合物Ganetespib可明显导致ALS1、ALS3、HWP1、BCR1、ACE2、RLM1下调和EAP1、ZAP1上调。
-
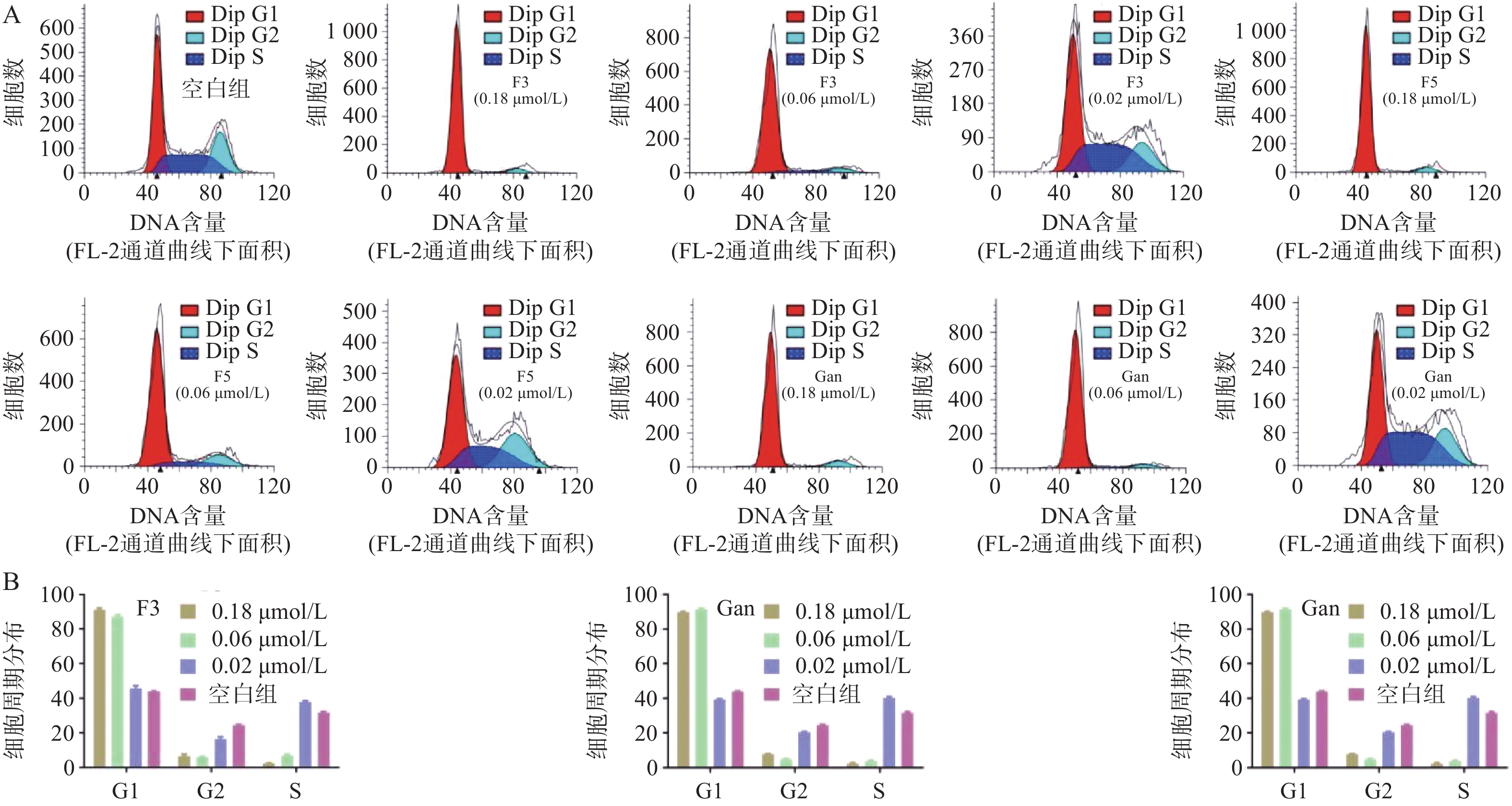
由于化合物F3和F5对血液肿瘤细胞系HEL相比于HL60和A549细胞具有更强的抑制作用,且血液肿瘤患者需使用抗真菌药物以预防IFIs。因此,以空白组作为对照,通过流式细胞术对化合物F3、F5以及先导化合物Ganetespib在多个浓度作用下HEL细胞的细胞周期分布进行了测定(图7A)。结果表明:化合物F3、F5以及Ganetespib均能够剂量依赖地阻滞HEL的细胞周期于G0/G1期;如化合物F3、F5以及Ganetespib在接近IC50的0.02 μmol/L的浓度下,处于G0/G1期的细胞占比分别为45.7%、45.3%和39.4%,与空白组的44.0%相当;但在0.06 μmol/L浓度下,G0/G1期的细胞占比分别明显地增加至87.2%、77.6%和91.5%;而在0.18 μmol/L浓度下,化合物F3和F5组分别进一步上升至91.7%和91.1%,Ganetespib组则略微下降至89.8%(图7B)。
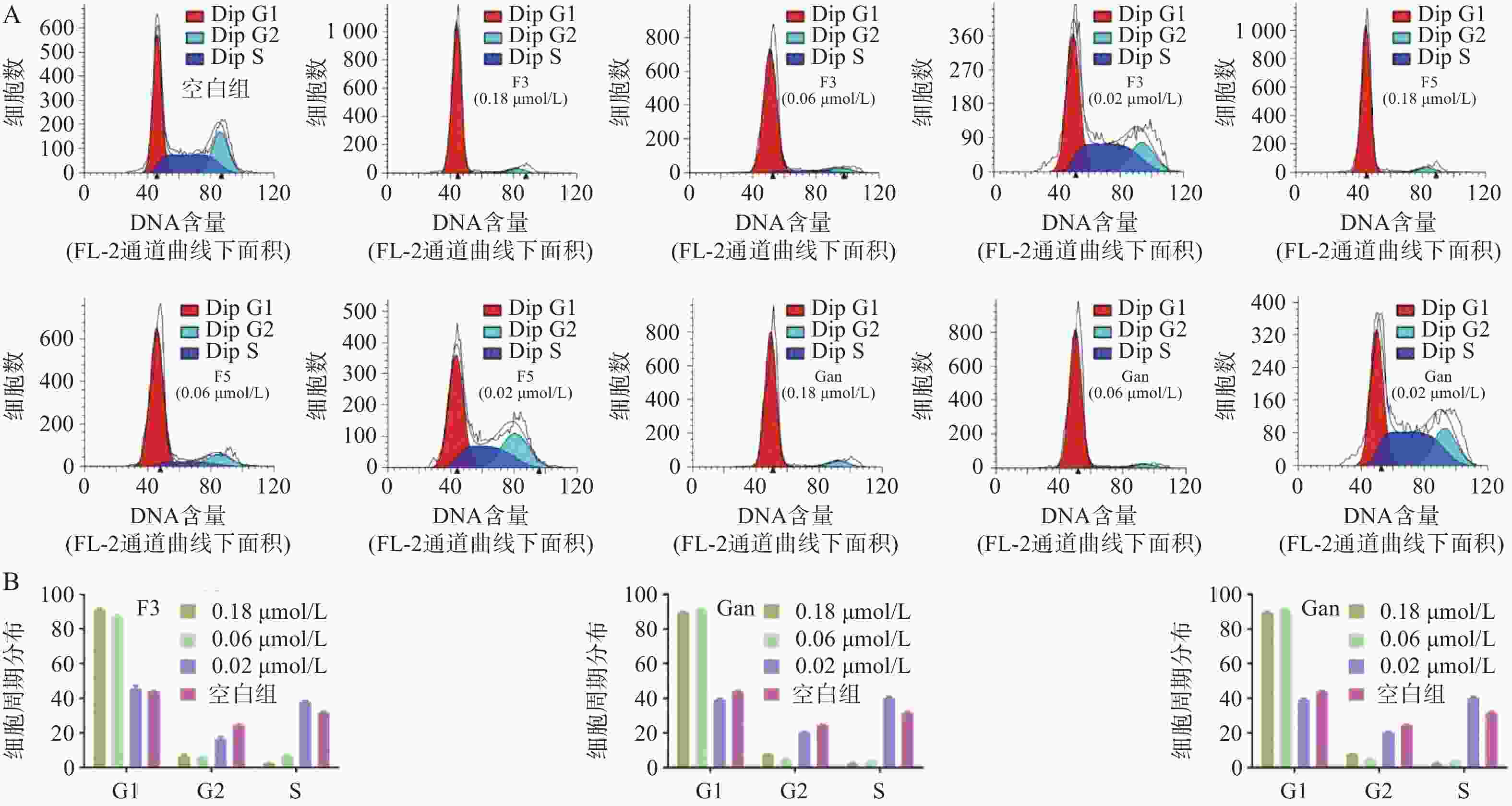
图 7 化合物作用下对细胞周期的影响
-
通过对前期发现的具有良好协同抗耐药真菌活性的Hsp90抑制剂Ganetespib进行结构改造,成功设计合成了19个新型Hsp90抑制剂,绝大部分化合物具有较强的Hsp90α抑制活性、良好的协同FLC抗耐药真菌活性及较优的抗肿瘤活性。其中,目标化合物F3和F5具有优秀的协同FLC抗耐药真菌活性(FICI均为0.047)和抗肿瘤活性(IC50分别为0.025~0.15 μmol/L,0.021~0.23 μmol/L)。机制研究表明,优选化合物F3和F5能下调真菌耐药基因和外排泵基因表达水平,抑制真菌生物被膜的形成;且能阻滞血液肿瘤细胞的细胞周期处于G0/G1期。研究结果提示,化合物F3和F5可以有效发挥协同FLC抗耐药真菌和抗肿瘤双重活性,这为开发具有抗耐药真菌和抗肿瘤双重作用的新药提供了新的思路。
Design, synthesis and antifungal and antitumor activity research of novel Hsp90 inhibitors
-
摘要:
目的 以Hsp90抑制剂Ganetespib为先导化合物,设计、合成兼具协同氟康唑(FLC)抗耐药真菌和抗肿瘤双重作用的新型Hsp90抑制剂。 方法 前期研究发现Ganetespib具有良好协同FLC抗耐药真菌活性,协同指数(FICI)= 0.023~0.039,本研究对Ganetespib进行结构改造,将其吲哚环替换为含不同取代基的苯环,设计合成一系列新化合物,并测定其体外协同FLC抗耐药真菌C. albicans 0304103和抗肿瘤(HEL、HL60和A549细胞)及Hsp90α抑制等活性,探讨其构效关系和作用机制。 结果 19个新化合物的化学结构经氢谱(1H NMR)、碳谱(13C NMR)和高分辨质谱(HRMS)确证;绝大多数化合物具有较强的Hsp90α抑制活性、良好的协同FLC抗耐药真菌活性和抗肿瘤活性,苯环上供电子基取代对提升协同FLC抗耐药真菌活性有利;其中,化合物F3和F5具有优秀的协同FLC抗耐药真菌活性(FICI均为0.047)和抗肿瘤活性(IC50分别为0.025~0.15 μmol/L,0.021~0.23 μmol/L),且能下调真菌耐药基因和外排泵基因表达水平,抑制真菌生物被膜的形成,并能阻滞HEL细胞的细胞周期于G0/G1期。 结论 新型Hsp90抑制剂如F3和F5均能有效发挥协同FLC抗耐药真菌和抗肿瘤双重活性,这为研发具有协同FLC抗耐药真菌和抗肿瘤双重作用的新药提供新的思路。 -
关键词:
- Hsp90抑制剂 /
- Ganetespib /
- 白念珠菌 /
- 抗真菌耐药性 /
- 抗肿瘤
Abstract:Objective To design and synthesize novel Hsp90 inhibitors with dual functions of synergistically enhancing the antifungal activity of fluconazole (FLC) against drug-resistant fungi and anti-tumor activity based on the Hsp90 inhibitor Ganetespib. Methods The previous research found that Ganetespib had a good synergistic anti-resistant fungal activity with FLC, with a fractional inhibitory concentration index (FICI) of 0.023 to 0.039. In this study, structural modifications were made to Ganetespib by replacing its indole ring with a phenyl ring containing different substituents to design and synthesize a series of new compounds. The in vitro synergistic anti-resistant fungal activity against C. albicans 0304103 in combination with FLC, anti-tumor activity (against HEL, HL60 and A549 cells), and Hsp90α inhibition activity were determined to explore their structure-activity relationship and mechanism of action. Results The chemical structures of 19 new compounds were confirmed by 1H NMR, 13C NMR and HRMS. Most of the compounds exhibited strong Hsp90α inhibitory activity, good synergistic activity against drug-resistant fungi in combination with FLC and anti-tumor activity. The substitution of electron-donating groups on the benzene ring was beneficial to enhancing the synergistic activity against drug-resistant fungi in combination with FLC. Among them, compounds F3 and F5 showed excellent synergistic activity against drug-resistant fungi in combination with FLC (FICI were both 0.047) and anti-tumor activity (IC50 were 0.025 to 0.15 μmol/L and 0.021 to 0.23 μmol/L respectively), and could down-regulate the expression levels of drug resistance genes and efflux pump genes in fungi, inhibit the formation of fungal biofilms, and arrest the cell cycle of HEL cells at G0/G1 phase. Conclusion The novel Hsp90 inhibitors such as F3 and F5 could both effectively exert the dual activities of synergizing with FLC to combat drug-resistant fungi and fight against tumors, which provided a new idea for the development of new drugs with dual functions of synergizing with FLC to combat drug-resistant fungi and fight against tumors. -
Key words:
- Hsp90 inhibitor /
- Ganetespib /
- Candida albicans /
- antifungal resistance /
- anti-tumor
-
图 3 目标化合物的合成路线
反应与条件:(a)PhOCOCl, CH2Cl2, Et3N, rt, 30 min, 产率75%~92%;(b)NH2NH2·H2O, EtOH, 回流, 3 h, 产率 72%~91%;(c)(i)EtOH, 1,4-dioxane, 回流, 2 h;(ii)K3Fe(CN)6, NaOH, 回流, 12 h 产率 53%~72%;(iii)(用于制备32o~32s)NaOH, H2O, 回流, 12 h, 产率 35%~49%;(d)H2, Pd/C, CH3OH, rt, 12 h, 产率 71%~85%。
图 5 化合物对真菌外排泵的抑制
A.化合物作用下C. albicans 0304103真菌耐药基因相对表达水平;B.不同时间点细胞外Rh6G荧光强度*P<0.05,**P<0.01,***P<0.001,与FLC(8 μg/ml)组比较。
图 6 化合物对真菌生物被膜和菌丝的抑制
A. 化合物作用下C. albicans 0304103的生物被膜形成率;B.化合物作用下C. albicans 0304103菌丝的倒置显微镜图;C.化合物作用下C. albicans 0304103生物被膜和菌丝形成相关基因的表达水平
表 1 Real time RT-PCR实验所用的基因引物序列
名称 序列 ERG11-F ACTCATGGGGTTGCCAATGT ERG11-R GAGCAGCATCACGTCTCCAA CDR1-F TCCACGGTCGTGAATTCCAATGTG CDR1-R GCCAGCAACAGGACCAGCTTC CDR2-F GCTACTGCCATGTCACTCTCCAC CDR2-R GGACAACTGTGCTTCCAGGAGTAG MDR1-F CCACTGGTGGTGCAAGTGTT MDR1-R TCGTTACCGGTGATGGCTCT ALS1-F GTGTCGGTTGTCAGAAGAGC ALS1-R TTGTTCACGTTGAGCCATGG ALS3-F ACTTTGTGGTCTACAACTTGGG ALS3-R CCAGATGGGGATTGTAAAGTGG HWP1-F CTGAACCTTCCCCAGTTGCT HWP1-R CGACAGCACTAGATTCCGGA EAP1-F TCCTACACGACTGACACTGC EAP1-R TGACACCCGTAGTTACTGCTG BCR1-F TCCTTTACGTGCACCACCTC BCR1-R ATGCCGACGATTCAGCTGAT ACE2-F ACTTTGTGGTCTACAACTTGGG ACE2-R CCAGATGGGGATTGTAAAGTGG RLM1-F GTGCCTGCGAATGTTCCAAA RLM1-R TGCATTGCTTCCTCCTGTCA ZAP1-F TACCGCGACTACAAACCACC ZAP1-R TGCCCCTGTTGCTCATGTTT ACT1-F GGTTTGGAAGCTGCTGGTAT ACT1-R ACCACCAATCCAGACAGAGT  下载: 导出CSV
下载: 导出CSV
表 2 化合物F1-F19的Hsp90α抑制活性、体外协同抗耐药真菌及抗肿瘤活性
化合物 取代基(R) Hsp90α C. albicans
0304103(μg/ml)HEL HL60 A549 抑制率(%,1 μmol/L) MIC80 FICI IC50(μmol/L) IC50(μmol/L) IC50(μmol/L) F1 H 99 >64 0.047 0.049±0.003 0.13±0.05 0.15±0.024 F2 4-OCH3 99 >64 0.047 0.026±0.003 0.17±0.014 0.32±0.031 F3 3-OCH3 97 >64 0.047 0.025±0.002 0.061±0.011 0.15±0.022 F4 2-OCH3 97 >64 0.063 0.048±0.007 0.069±0.005 0.55±0.15 F5 4-CH3 97 >64 0.047 0.021±0.002 0.067±0.005 0.23±0.026 F6 3-CH3 97 >64 0.047 0.052±0.002 0.12±0.017 0.22±0.02 F7 2-CH3 98 >64 0.023 0.107±0.019 0.28±0.029 0.79±0.076 F8 4-F 95 >64 0.125 0.138±0.012 0.325±0.026 0.26±0.008 F9 3-F 97 >64 0.063 0.115±0.012 0.25±0.008 0.25±0.063 F10 2-F 95 >64 0.047 0.037±0.002 0.083±0.004 0.40±0.067 F11 2,4-2F 94 >64 0.188 0.153±0.012 0.466±0.012 0.76±0.012 F12 4-CF3 96 >64 2 0.033±0.001 0.115±0.007 0.10±0.022 F13 3-CF3 97 >64 0.094 0.085±0.009 0.361±0.024 0.47±0.1 F14 2-CF3 98 >64 − 0.408±0.022 0.650±0.014 0.22±0.02 F15 4-NH2 99 >64 0.063 0.047±0.003 0.055±0.003 0.65±0.14 F16 3-NH2 97 >64 0.125 0.070±0.034 0.077±0.006 0.25±0.063 F17 2-NH2 96 >64 0.141 0.120±0.025 0.135±0.013 0.63±0.068 F18 4-CO2H 94 >64 − 5.5±0.73 16±2.9 43±3 F19 3-CO2H 95 >64 − 16±1.2 13±0.47 21±2.1 Ganetespib − 99 >64 0.023 0.021±0.002 0.023±0.002 0.11±0.019
下载: 导出CSV
-
[1] AHMADI N, AHMADI A, KHEIRALI E, et al. Systemic infection with Candida albicans in breast tumor bearing mice: Cytokines dysregulation and induction of regulatory T cells[J]. J Mycol Med, 2019, 29(1):49-55. doi: 10.1016/j.mycmed.2018.10.006 [2] LI J, BUCHNER J. Structure, function and regulation of the hsp90 machinery[J]. Biomed J, 2013, 36(3):106-117. doi: 10.4103/2319-4170.113230 [3] LIN S F, LIN J D, HSUEH C, et al. Efficacy of an HSP90 inhibitor, ganetespib, in preclinical thyroid cancer models[J]. Oncotarget, 2017, 8(25):41294-41304. doi: 10.18632/oncotarget.17180 [4] WANG M N, SHEN A J, ZHANG C, et al. Development of heat shock protein(Hsp90)inhibitors to combat resistance to tyrosine kinase inhibitors through Hsp90-kinase interactions[J]. J Med Chem, 2016, 59(12):5563-5586. doi: 10.1021/acs.jmedchem.5b01106 [5] COWEN L E. The fungal Achilles' heel: targeting Hsp90 to cripple fungal pathogens[J]. Curr Opin Microbiol, 2013, 16(4):377-384. doi: 10.1016/j.mib.2013.03.005 [6] TIWARI S, THAKUR R, SHANKAR J. Role of heat-shock proteins in cellular function and in the biology of fungi[J]. Biotechnol Res Int, 2015, 2015:132635. [7] LI L P, AN M M, SHEN H, et al. The non-Geldanamycin Hsp90 inhibitors enhanced the antifungal activity of fluconazole[J]. Am J Transl Res, 2015, 7(12):2589-2602. [8] YUAN R, TU J, SHENG C Q, et al. Effects of Hsp90 inhibitor ganetespib on inhibition of azole-resistant Candida albicans[J]. Front Microbiol, 2021, 12:680382. doi: 10.3389/fmicb.2021.680382 [9] HE S, DONG G, WU S, et al. Small molecules simultaneously inhibiting p53-murine double minute 2(MDM2)interaction and histone deacetylases(HDACs): discovery of novel multitargeting antitumor agents[J]. J Med Chem, 2018, 61(16):7245-7260. doi: 10.1021/acs.jmedchem.8b00664 [10] WHITESELL L, ROBBINS N, HUANG D S, et al. Structural basis for species-selective targeting of Hsp90 in a pathogenic fungus[J]. Nat Commun, 2019, 10(1):402. doi: 10.1038/s41467-018-08248-w [11] ROUDBARMOHAMMADI S, ROUDBARY M, BAKHSHI B, et al. ALS1 and ALS3 gene expression and biofilm formation in Candida albicans isolated from vulvovaginal candidiasis[J]. Adv Biomed Res, 2016, 5:105. doi: 10.4103/2277-9175.183666 [12] NOBILE C J, NETT J E, ANDES D R, et al. Function of Candida albicans adhesin Hwp1 in biofilm formation[J]. Eukaryot Cell, 2006, 5(10):1604-1610. doi: 10.1128/EC.00194-06 [13] LI F, SVAROVSKY M J, KARLSSON A J, et al. Eap1p, an adhesin that mediates Candida albicans biofilm formation in vitro and in vivo[J]. Eukaryot Cell, 2007, 6(6):931-939. doi: 10.1128/EC.00049-07 [14] NOBILE C J, ANDES D R, NETT J E, et al. Critical role of Bcr1-dependent adhesins in C. albicans biofilm formation in vitro and in vivo[J]. PLoS Pathog, 2006, 2(7):e63. doi: 10.1371/journal.ppat.0020063 [15] KELLY M T, MACCALLUM D M, CLANCY S D, et al. The Candida albicans CaACE2 gene affects morphogenesis, adherence and virulence[J]. Mol Microbiol, 2004, 53(3):969-983. doi: 10.1111/j.1365-2958.2004.04185.x [16] OLIVEIRA-PACHECO J, ALVES R, COSTA-BARBOSA A, et al. The role of Candida albicans transcription factor RLM1 in response to carbon adaptation[J]. Front Microbiol, 2018, 9:1127. doi: 10.3389/fmicb.2018.01127 [17] NOBILE C J, NETT J E, HERNDAY A D, et al. Biofilm matrix regulation by Candida albicans Zap1[J]. PLoS Biol, 2009, 7(6):e1000133. doi: 10.1371/journal.pbio.1000133 [18] 郭东东, 岳慧珍, 魏羽佳, 黄广华. 白念珠菌生物被膜形成的遗传调控机制[J]. 生物工程学报, 2017, 33(9):1567-1581. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 14546
- HTML全文浏览量: 4160
- PDF下载量: 35
- 被引次数: 0
