

 下载:
下载:





-
卡培他滨是一种新型的氟尿嘧啶类口服药,它是5-氟尿嘧啶(5-FU)的前体药物。卡培他滨口服给药方便、依从性好、疗效确切,是治疗结直肠癌的基石药物,但在治疗过程中产生的不良反应(手足综合征等)极大地影响了患者的治疗。手足综合征(HFS)是卡培他滨引起的不良反应中较为特殊的反应,也是卡培他滨在治疗过程中的药物剂量限制性不良反应,主要表现为皮肤红肿、水泡、出血、疼痛。合并使用环氧合酶、尿素霜、维生素B6、奥美拉唑也不能完全阻断手足综合征的发生[1-3];合并使用中草药如白芍、桂枝、甘草等可降低手足综合征的发生率[4]。口服卡培他滨的患者手足综合征发生率高达60%[1],严重降低患者用药依从性,严重手足综合征(约17%)的患者只能减少药物摄入量乃至停止服用药物,影响化疗按期、足量进行[5-6]。明确手足综合征的发生机制和有效干预对卡培他滨安全有效的应用具有重要价值。目前手足综合征模型的建立还没有统一的金标准,本实验尝试用ICR小鼠灌胃卡培他滨后建立手足综合征模型,为卡培他滨致手足综合征模型的建立及机制研究提供参考。
-
雄性ICR小鼠,SPF级,5周龄左右,共42只小鼠,小鼠由上海西普尔-必凯实验动物有限公司提供。经上海中医药大学动物伦理委员会批准,实验动物伦理审查号为PZSHUTCM210715004。将小鼠随机编号1~42号,对照组ICR小鼠6只,实验组ICR小鼠36只。卡培他滨(商品名希罗达,上海罗氏制药有限公司,规格:0.5 g/片,国药准字号H20073024);羧甲基纤维素钠(CMC-Na)(国药集团);离心管(Titan公司);动物解剖手术器材(瑞沃德公司);消毒酒精、棉花球、锡箔纸(国药集团)。
-
造模前小鼠先饲养1周,保证充足的水源与饲料,42只ICR小鼠自由摄取食物,环境湿度保持区间50%~60%(±5%),控制温度在(23±1) ℃,调节光照为12 h(6: 30至18: 30光照),其余时间为光暗周期。定期观察小鼠培养环境,定期为小鼠更换垫料,清理排泄物。
-
卡培他滨0.5 g/片,溶解于10 ml CMC-Na(0.5%)中,制得50 mg/ml悬浊液备用。实验组ICR小鼠(36只),灌胃给药,按275 mg/kg(即小鼠0.15 ml/30g)连续灌胃14 d,2次/d(按照卡培他滨临床给药方案1 250 mg/kg,2次/d的剂量换算)。对照组ICR小鼠(6只),灌胃给予CMC-Na(0.5%)溶液,按照4 ml/kg剂量连续的灌胃14 d,2次/d。
-
取ICR小鼠的后足跖部皮肤组织,用生理盐水冲洗并擦干,以多聚甲醛(4%)对小鼠组织进行固定,石蜡包埋切片,使用苏木精-伊红对小鼠组织染色(H&E染色),使用普通光镜(比例尺5 μm)检测(注:染色呈现蓝色的为表皮层,该层位于淡粉色的角质层和淡蓝色的真皮层之间)。每天肉眼观察,若小鼠出现足跖部特征性改变,立即拍照并记录手足综合征发生时间,每3 d拍照记录小鼠足跖部皮肤变化情况。
-
实验组与对照组自由饲养1周后称重,记录原始体重,灌胃给药后每3 d对所有实验小鼠称重一次并记录体重变化。
-
标准参考美国国家癌症研究所(NCI)关于手足综合征的分级规定,建模实验中若实验小鼠足跖部的皮肤呈现了红色斑块、组织肿胀、角质层脱屑、溃疡、角质层厚度明显增加及其他明显体征者均可判定为手足综合征阳性。
-
对于手足综合征阳性的小鼠,在出现手足综合征后,经眼眶采样,用1.5 ml离心管(肝素钠抗凝)取血约0.5 ml,12 000 r/min离心10 min,收集血浆约0.25 ml,置于−80 ℃冰箱冷冻保存。采集小鼠血样后按照实验动物伦理要求规范处死小鼠,用手术剪取小鼠手足部皮肤,生理盐水冲洗后用锡纸包裹住样本并标号。所有样本置于−80 ℃冰箱冷冻保存,所有操作在1 h内完成。
-
小鼠经过14 d给药未出现手足综合征阳性反应,则认定该类小鼠为造模失败小鼠,在第15天对该类小鼠统一进行眼眶采血和足趾部皮肤组织样本收集,方法同“1.5.1”。对照组小鼠在第15天收集样本,方法同“1.5.1”。
-
数据采用平均值±标准差(mean±SD)表示,组与组之间根据数据分布状态比较采用学生t检验或秩检验,P<0.05认为有统计学差异。制图采用Excel或Graphpad 8.0软件。
-
通过对比对照组与实验组ICR小鼠体重变化,发现对照组ICR小鼠,灌胃14 d后,体重明显增加;而实验组ICR小鼠,灌胃14 d后,体重明显降低。表1和图1 为14 d内ICR小鼠的体重变化,实验过程中实验组小鼠死亡3只。
表 1 实验周期内小鼠体重变化情况(m/g,
$ \bar{x}\pm s $ )组别 n 0 d 7 d 14 d 对照组 6 28.54±0.71 32.56±0.88 34.66±1.07 实验组 33 29.07±0.76 26.84±1.74*** 23.15±2.31*** *** P<0.001,与对照组比较。 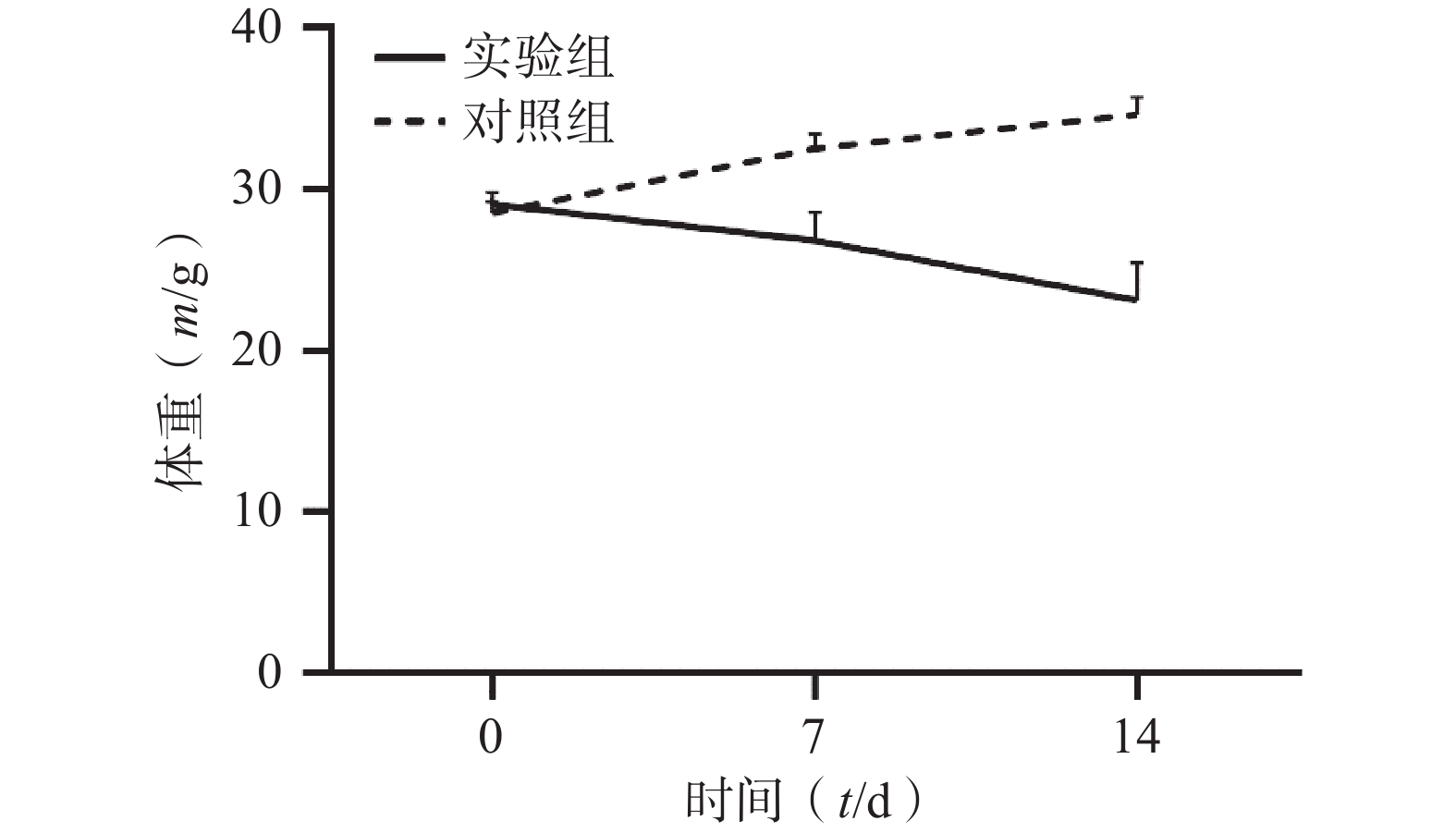
图 1 ICR小鼠体重变化折线图
-
与对照组ICR小鼠相比,19只实验组小鼠的足底皮肤明显出现红斑、肿涨并出现少许水泡(图2),认为该小鼠出现手足综合征,按照伦理要求将小鼠处死,收集足底皮肤组织样本和血浆样本。

图 2 小鼠足底皮肤图像
-
将ICR小鼠四肢皮肤用4%多聚甲醛固定后进行H&E染色(因苏木精呈碱性,细胞核内的染色质与胞质内的核酸显紫蓝色;伊红呈酸性,细胞质和细胞外基质中的成分显红色),如图3显示。与正常ICR小鼠相比,手足综合征阳性小鼠皮肤表皮层增厚,角质层呈现粉红色,真皮层呈现浅蓝色,表皮层处在二者之间呈现蓝色。发生HFS小鼠皮肤颗粒层变薄,基底层和棘层间细胞数量减少。正常小鼠角质层明显较薄。
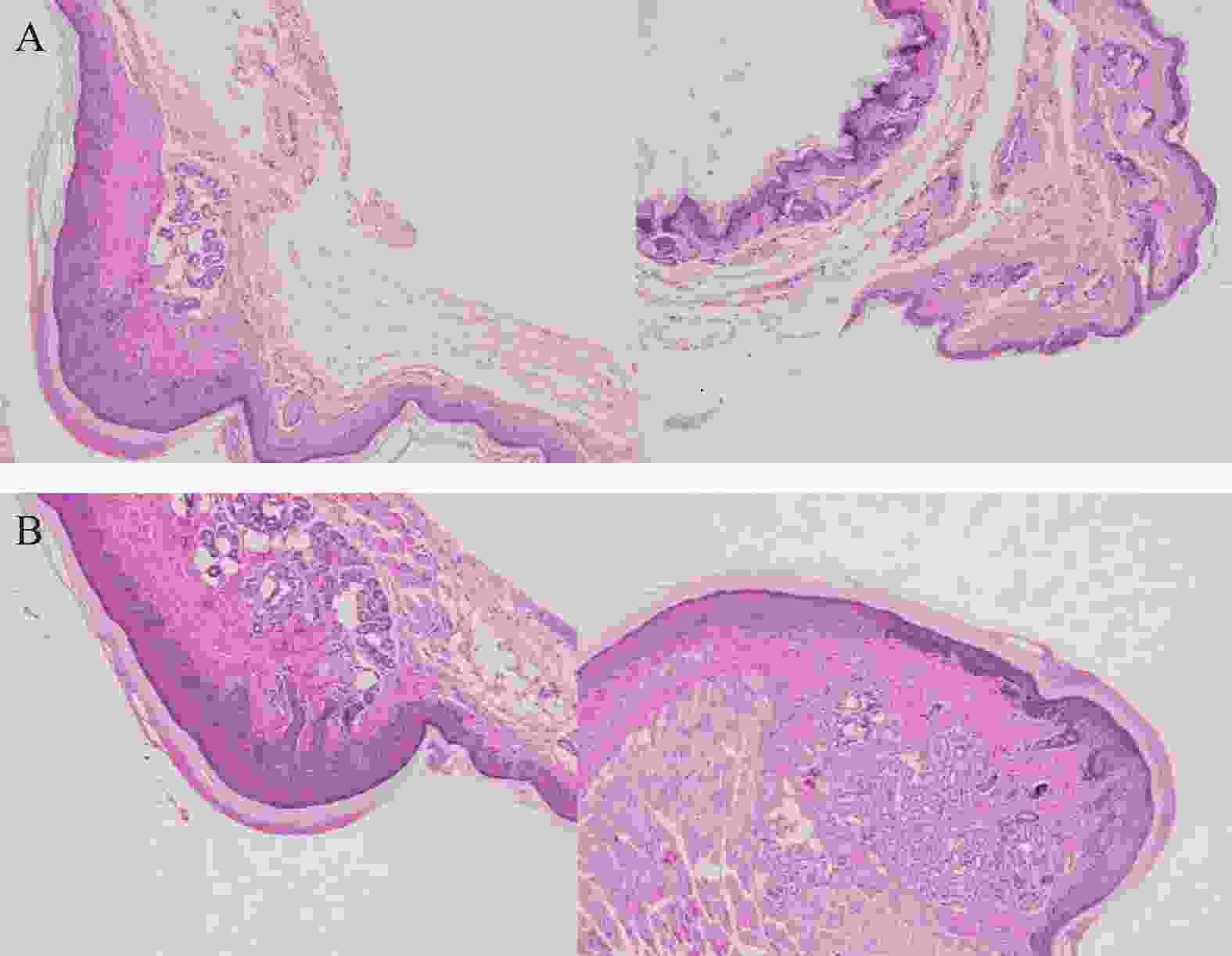
图 3 足底皮肤组织H&E染色图(4×10倍)
-
ICR小鼠共39只(对照组6只,实验组33只),19只实验组ICR小鼠出现手足综合征阳性症状,发生率为57.58%。实验组ICR小鼠灌胃给药1周,出现手足综合征阳性小鼠与未出现手足综合征小鼠相比:实验组小鼠足底皮肤颜色变深(深红色),少量小鼠四肢掌心有透明水泡样组织出现;在灌胃给药10 d后,实验组小鼠出现手足综合征阳性症状,四肢出现了红斑、脱屑、水泡(红斑出现最多,脱屑其次,水泡最少)等情况。
-
将收集的血浆样本按照前期报道的方法进行卡培他滨及其5种代谢产物(5'-脱氧-5-氟胞嘧啶核苷、去氧氟尿苷、5'-氟-2'-脱氧尿苷、5-氟尿嘧啶、5-氟二氢嘧啶-2,4-二酮[7])定量(ng/ml,mean±SD)。结果发现,发生手足综合征小鼠与未发生手足综合征小鼠相比,卡培他滨浓度(ng/ml)分别为(58.08±44.54)和(39.23±26.98),5'-脱氧-5-氟胞嘧啶核苷浓度(ng/ml)分别为(
6047.42 ±3331.94 )和(4442.77 ±2140.44 ),去氧氟尿苷浓度(ng/ml)分别为(2899.28 ±1821.15 )和(2018.81 ±1037.86 ),5'-氟-2'-脱氧尿苷浓度(ng/ml)分别为(112.89±36.85) 和(122.23±19.16),5-氟尿嘧啶浓度(ng/ml)分别为(46.86±23.08)和(38.33±20.62),5-氟二氢嘧啶-2,4-二酮浓度(ng/ml)分别为(24.45±14.79)和(27.34±17.84)。卡培他滨及其代谢产物在发生和未发生手足综合征小鼠体内暴露水平均无明显差异(P>0.05,图4)。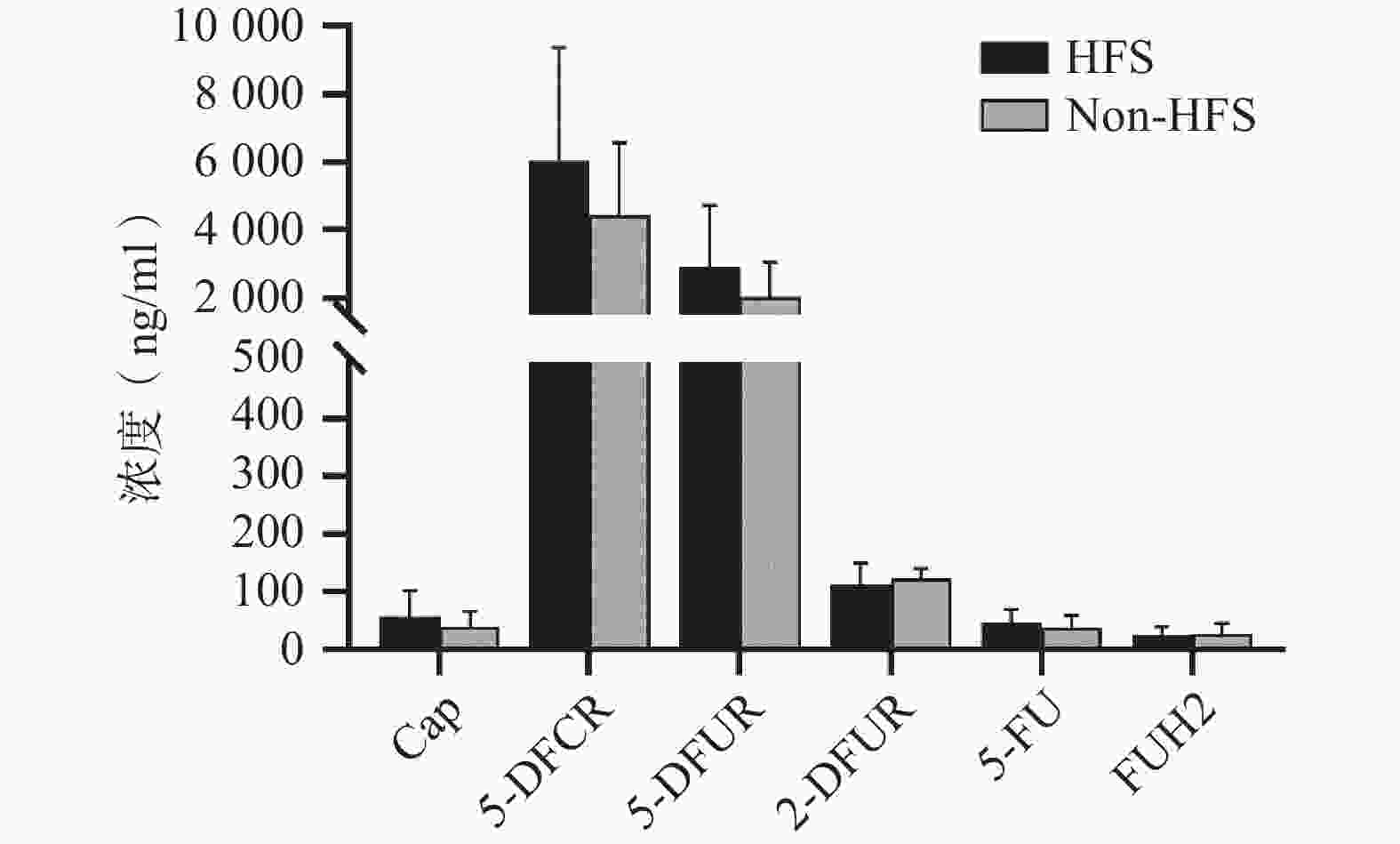
图 4 卡培他滨及其代谢产物暴露浓度分析
-
体内药物暴露水平同药物疗效和不良反应密切相关。目前药物暴露水平常用的参数有曲线下面积(AUC)、稳态浓度(css)、峰浓度(cmax)、谷浓度(cmin)等。有研究发现[8],根据5-FU的AUC调整给药剂量后,患者血液系统出现3~4级不良反应发生率从之前的17.5%下降为 7.6%(P<0.05),黏膜炎的发生率也从5%降低为0%(P<0.01),总生存时间显著增加,从原来的16个月延长到22个月(P<0.01)。因此,5-FU在体内浓度可能成为预测疗效及不良反应的潜在预警生物标志物之一[9-10]。本研究中未发现卡培他滨及其代谢产物同手足综合征具有相关性,但Daher Abdi在20名75岁以上老年患者中发现,卡培他滨及其代谢产物在发生手足综合征患者体内暴露水平明显较高[11],但体外研究表明,5-FU同手足综合征无明显关联[12]。另有研究表明,手足皮肤局部较高表达的胸苷磷酸化酶可产生局部较高浓度的5-FU,同卡培他滨引发的手足综合征密切相关[13]。总之,卡培他滨引发手足综合征的机制仍需进一步研究。
在卡培他滨致手足综合征模型的建立方面,黎鹏等灌胃给予SD大鼠200~400 mg/d,2次/d,用药1~2周(1周后停药3 d),建立了卡培他滨致手足综合征模型,并认为200 mg/kg的剂量,灌胃2周可较好地建立手足综合征模型(造模成功率77.5%)[14]。但有研究显示,卡培他滨在大鼠体内的代谢过程与人体不同,因大鼠体内胞苷脱胺酶活性较低,5-DFCR通过糖基化生成其他产物,产生的5-FU较少,而卡培他滨在小鼠体内的代谢过程与人体基本相同,可能是最好的动物模型[15-16];Hiromoto [3]等利用ICR小鼠,灌胃给予200 mg/kg,1次/d,5次/周的方法建立手足综合征模型,3周建立手足综合征模型,但成功率未报道。He和Chen等给予ICR小鼠灌胃200 mg/kg,1次/d,连续灌胃30 d后,6只小鼠中仅有1只未发生手足综合征[17]。前期预实验中,采用灌胃200 mg/kg,2次/d的方法在ICR小鼠上建立手足综合征模型,灌胃1周后,6只小鼠仅有1只发生手足综合征。因此,根据临床卡培他滨用药剂量(1 250 mg/kg,2次/d)换算小鼠剂量为275 mg/kg,2次/d,既缩短了给药时间,又提高了建模成功率(57.6%),且较少有小鼠死亡(3只)。
-
本实验成功建立了卡培他滨致手足综合征的ICR小鼠模型,在给药时长、造模成功率方面均有一定优势。卡培他滨及其代谢产物在小鼠体内暴露浓度同手足综合征发生可能无关。手足综合征的发生机制仍需进一步研究。
Establishment of mouse model of hand-foot syndrome induced by capecitabine
-
摘要:
目的 手足综合征是卡培他滨的剂量限制性毒性反应。目前手足综合征模型的建立并没有统一的金标准,本实验给予ICR小鼠灌胃卡培他滨,诱导手足综合征发生,为手足综合征模型的建立提供借签。 方法 42只雄性ICR小鼠随机分为对照组(6只)和实验组(36只),实验组持续2周灌胃给予卡培他滨(275 mg/kg,2次/d),对照组给予溶剂0.5% CMC-Na(4 ml/kg,2次/d),取小鼠足部皮肤样本进行H&E染色,观察足跖部特征性外观以及形态改变,评估手足综合征的动物模型是否成功构建;实验结束后处死小鼠,收集血浆,定量其中卡培他滨及其代谢产物浓度改变。 结果 6只对照组小鼠均未出现手足综合征症状;实验组19只小鼠足部皮肤出现红斑、肿胀等症状,H&E染色可见部分足底皮肤角表皮层增厚,部分角质脱失破损,判断为发生手足综合征。发生与未发生手足综合征小鼠血浆中卡培他滨及其代谢产物浓度未见明显差异。 结论 卡培他滨致小鼠手足综合征的模型构建成功;卡培他滨及其代谢产物体内暴露水平差异可能不是手足综合征发生的原因。 Abstract:Objective Hand-foot syndrome is a dose-limiting toxicity of capecitabine. At present, there is no unified gold standard for the establishment of hand-foot syndrome model. To induce hand-foot syndrome and provide a reference for the establishment of hand-foot syndrome model by administering capecitabine in ICR mice. Methods 42 male ICR mice were randomly divided into control group (6 mice) and experimental group (36 mice). The experimental group was given capecitabine (275 mg/kg, twice/d) by intragastric administration for two weeks, and the control group was given 0.5% CMC-Na (4 ml/kg, twice/d), to evaluate whether the animal model of hand-foot syndrome was successfully constructed through H&E staining of mouse foot skin samples and observe morphological changes and the characteristic appearance of mouse foot skin. After the experiment, the mice were sacrificed, and plasma was collected to quantify the concentrations of capecitabine and metabolites. Results Control mice did not showed symptoms of hand-foot syndrome. The skin of the feet of 19 mice in the experimental group showed symptoms such as erythema and swelling, and H&E staining results showed that the plantar skin angular epidermis was thickened, and part of the keratin was exfoliated and damaged, which was considered to be hand-foot syndrome. There were no significant differences in the concentrations of capecitabine and its metabolites between mice with and without hand-foot syndrome. Conclusion The model of hand-foot syndrome induced by capecitabine in mice was successfully established. Differences in exposure levels of capecitabine and metabolites may not be the cause of hand-foot syndrome. -
Key words:
- capecitabine /
- hand-foot syndrome /
- animal model /
- drug concentration /
- ICR mice
-
图 4 卡培他滨及其代谢产物暴露浓度分析
注:Cap:卡培他滨;5-DFCR:5'-脱氧-5-氟胞嘧啶核苷;5-DFUR:去氧氟尿苷;2-DFUR:5'-氟-2'-脱氧尿苷;5-FU:5-氟尿嘧啶;FUH2:5-氟二氢嘧啶-2,4-二酮。
表 1 实验周期内小鼠体重变化情况(m/g,
$ \bar{x}\pm s $ )组别 n 0 d 7 d 14 d 对照组 6 28.54±0.71 32.56±0.88 34.66±1.07 实验组 33 29.07±0.76 26.84±1.74*** 23.15±2.31*** *** P<0.001,与对照组比较。  下载: 导出CSV
下载: 导出CSV
-
[1] PANDY J G P, FRANCO P I G, LI R K. Prophylactic strategies for hand-foot syndrome/skin reaction associated with systemic cancer treatment: a meta-analysis of randomized controlled trials[J]. Support Care Cancer, 2022, 30(11):8655-8666. doi: 10.1007/s00520-022-07175-3 [2] BRAIK T, YIM B, EVANS A, et al. Randomized trial of vitamin B6 for preventing hand-foot syndrome from capecitabine chemotherapy[J]. J Community Support Oncol, 2014, 12(2):65-70. doi: 10.12788/jcso.0017 [3] HIROMOTO S, KAWASHIRI T, YAMANAKA N, et al. Use of omeprazole, the proton pump inhibitor, as a potential therapy for the capecitabine-induced hand-foot syndrome[J]. Sci Rep, 2021, 11(1):8964. doi: 10.1038/s41598-021-88460-9 [4] DENG B, SUN W. Herbal medicine for hand-foot syndrome induced by fluoropyrimidines: a systematic review and meta-analysis[J]. Phytother Res, 2018, 32(7):1211-1228. doi: 10.1002/ptr.6068 [5] CARONIA D, MARTIN M, SASTRE J, et al. A polymorphism in the cytidine deaminase promoter predicts severe capecitabine-induced hand-foot syndrome[J]. Clin Cancer Res, 2011, 17(7):2006-2013. doi: 10.1158/1078-0432.CCR-10-1741 [6] 王志鹏. 卡培他滨不良反应和耐药生物标志物的发现[D]. 上海: 中国人民解放军海军军医大学, 2019. [7] WANG Z P, LI X X, YANG Y, et al. A sensitive and efficient method for determination of capecitabine and its five metabolites in human plasma based on one-step liquid-liquid extraction[J]. J Anal Methods Chem, 2019, 2019:1-10. [8] FETY R, ROLLAND F, Barberi-Heyob M, et al. Clinical impact of pharmacokinetically-guided dose adaptation of 5-fluorouracil: results from a multicentric randomized trial in patients with locally advanced head and neck carcinomas. Clin Cancer Res. 1998, 4(9): 2039-2045. [9] GAMELIN E, DELVA R, JACOB J, et al. Individual fluorouracil dose adjustment based on pharmacokinetic follow-up compared with conventional dosage: results of a multicenter randomized trial of patients with metastatic colorectal cancer. J Clin Oncol. 2008, 26(13): 2099-105. [10] MEZA-JUNCO J, SAWYER M B. Drug exposure: still an excellent biomarker[J]. Biomark Med, 2009, 3(6):723-731. doi: 10.2217/bmm.09.58 [11] DAHER ABDI Z, LAVAU-DENES S, PRÉMAUD A, et al. Pharmacokinetics and exposure-effect relationships of capecitabine in elderly patients with breast or colorectal cancer[J]. Cancer Chemother Pharmacol, 2014, 73(6):1285-1293. doi: 10.1007/s00280-014-2466-0 [12] FISCHEL J L, FORMENTO P, CICCOLINI J, et al. Lack of contribution of dihydrofluorouracil and α-fluoro-β-alanine to the cytotoxicity of 5'-deoxy-5-fluorouridine on human keratinocytes[J]. Anti Cancer Drugs, 2004, 15(10):969-974. doi: 10.1097/00001813-200411000-00006 [13] YANG B X, XIE X R, LV D Z, et al. Capecitabine induces hand-foot syndrome through elevated thymidine phosphorylase-mediated locoregional toxicity and GSDME-driven pyroptosis that can be relieved by tipiracil[J]. Br J Cancer, 2023, 128(2):219-231. doi: 10.1038/s41416-022-02039-3 [14] 黎鹏, 王炳胜, 李永民. 卡培他滨诱导手足综合征动物模型的建立[J]. 中华肿瘤防治杂志, 2017, 24(12):802-807. [15] DESMOULIN F, GILARD V, MALET-MARTINO M, et al. Metabolism of capecitabine, an oral fluorouracil prodrug: 19F NMR studies in animal models and human urine[J]. Drug Metab Dispos, 2002, 30(11):1221-1229. doi: 10.1124/dmd.30.11.1221 [16] DESMOULIN F, GILARD V, MARTINO R, et al. Isolation of an unknown metabolite of capecitabine, an oral 5-fluorouracil prodrug, and its identification by nuclear magnetic resonance and liquid chromatography–tandem mass spectrometry as a glucuroconjugate of 5’-deoxy-5-fluorocytidine, namely 2’-(β-d-glucuronic acid)–5’-deoxy-5-fluorocytidine[J]. J Chromatogr B, 2003, 792(2):323-332. doi: 10.1016/S1570-0232(03)00319-2 [17] HE X Y, WANG J L, WANG Q, et al. P38 MAPK, NF-κB, and JAK-STAT3 signaling pathways involved in capecitabine-induced hand-foot syndrome via interleukin 6 or interleukin 8 abnormal expression[J]. Chem Res Toxicol, 2022, 35(3):422-430. doi: 10.1021/acs.chemrestox.1c00317 -

 点击查看大图
点击查看大图

图(4) / 表(1)
计量
- 文章访问数: 13858
- HTML全文浏览量: 4797
- PDF下载量: 42
- 被引次数: 0
