

 下载:
下载:

-
络塞维(rosavin)是从中国药用植物红景天的根和茎中提取的独特化学成分[1],主要用于抗疲劳、抗缺氧、缓解压力、降血糖、提高工作能力效率和治疗神经系统的功能性疾病[2-3]。传统的络塞维获取方法主要是从红景天植物中分离提纯得到,但由于络塞维在红景天中的含量极低,且分离难度大,由于原材料的差异,含量也各有不同。鉴于天然络塞维的获取难度极大,而且价格昂贵,因此,开发一种廉价、简单、高收率的络塞维合成方法具有重大意义。本实验以廉价的葡萄糖、阿拉伯吡喃糖、肉桂醇为起始原料,经合成络思为其中一个中间体[4],然后,对络思葡萄糖6位羟基以叔丁基二苯基硅基进行高选择性保护,再乙酰化保护2,3,4位羟基,选择性脱去6位叔丁基二苯基硅基作为糖基化受体,三氟甲磺酸三甲基硅酯为糖苷化催化剂,1-O-三氯亚胺酯三乙酰阿拉伯糖为供体,实现了络塞维的全合成,合成路线如图1所示。
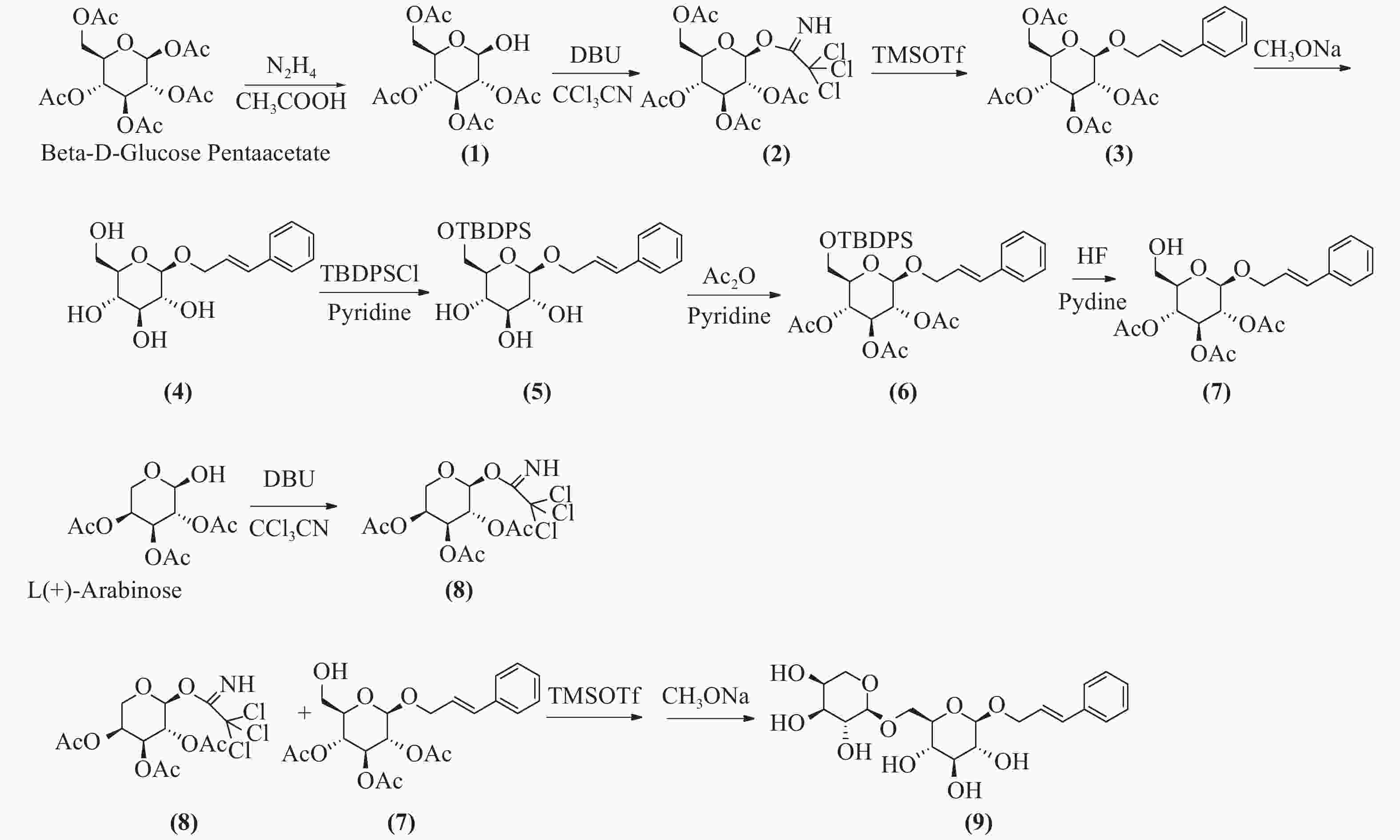
图 1 络塞维的全合成
-
β-D-五乙酰葡萄糖、四乙酰阿拉伯糖、肉桂醇、三氟甲磺酸三甲基硅脂、水合肼、甲醇钠等试剂均为分析纯,购买于上海阿达玛斯试剂有限公司。旋转蒸发仪、磁力搅拌器(德国艾卡仪器设备有限公司);低温冷阱(上海豫康科技有限公司);天平(梅特勒-托利多仪器有限公司);AC-P600型核磁共振仪(Bruker-Spectrospin);LC/MSD-iQ型质谱仪(安捷伦)。
-
将β-D-五乙酰葡萄糖780 g(2 mol)加入到800 ml的二甲基甲酰胺(DMF)中,加入乙酸133 g(2.2 mol),冷却至0 ℃,滴加水合肼110 g(2.2 mol),滴加完毕后,升至室温反应,用薄层色谱法(TLC)检测反应进行程度,反应完毕后,加入1 mol/L盐酸300 ml淬灭反应,乙酸乙酯(EA)萃取,饱和碳酸钠洗涤,干燥后蒸干,得无色油状物,快速硅胶柱层析得化合物(1)640 g,收率92%。1H-NMR (600 MHz, DMSO-d6, δ) 7.28 (d, J = 4.3 Hz, 1H), 5.47~5.33(m, 1H), 5.24 (t, J = 3.3 Hz, 1H), 4.94 (t, J = 9.6 Hz, 1H), 4.73 (dd, J = 10.3、3.4 Hz, 1H), 4.15 (dd, J = 8.5, 2.4 Hz, 2H), 4.04 (dd, J = 15.5, 8.5 Hz, 1H), 2.03 (s, 3H), 2.01 (d, J = 1.4 Hz, 3H), 1.98 (s, 3H)。13C -NMR (151 MHz, DMSO-d6, δ) 170.54, 170.17, 170.09 ~ 170.00 (4 C), 169.78, 89.50, 71.29, 69.85, 68.98, 66.76, 62.61, 20.88. ESI-MS C14H20O10 Na [M+Na]+ m/z=371.0951。
-
将四乙酰葡萄糖216 g(620 mmol)溶于350 ml无水二氯甲烷中,加入三氯乙腈160 g(1117 mmol),搅拌均匀,冷却至0 ℃,滴加1,8-二氮杂二环十一碳-7-烯(1,8-diazabicyclo [5.4.0] undec-7-ene, DBU)9.4 g(62 mmol),升至室温,反应3 h,TLC检测反应完毕,蒸干,石油醚:乙酸乙酯(2:1)快速硅胶柱层析,得无色油状化合物(2)265 g,收率87%。1H-NMR (CDCl3, δ): 6.60 (1H, d, J = 3.2 Hz, H-1), 5.54 (1H, t, J = 9.3 Hz, H-4), 5.30~5.10 (2H, 宽峰, H-2, H-3), 4.30~4.00 (3H, m, H-5, H2-6), 2.14~ 1.95 (12H, COCH3)。13C-NMR (300 MHz, DMSO-d6, δ) 170.5, 169.8, 169.7, 169.6, 159.7, 90.5, 71.2 (2 C), 67.8, 68.8, 65.4, 62.0, 21.0, 20.7, 20.65, 20.6。ESI-MS C16H20Cl3NO10Na [M+Na]+ m/z=514.0065。
-
将化合物(2)170 g(346 mmol)溶于1 L无水二氯甲烷中,加入肉桂醇70 g(520 mmol),搅拌下加入4 Å分子筛100 g,室温下搅拌30 min,然后冷却至0 ℃,滴加三氟甲磺酸三甲基硅8 g(34.6 mmol),室温搅拌反应过夜,TLC检测反应完毕后,过滤,滤液加饱和碳酸氢钠水溶液洗涤,饱和氯化钠洗涤,无水硫酸钠干燥,蒸干,快速硅胶柱层析得化合物(2R,3R,4S,5R,6R)-2-(acetoxymethyl)-6-(cinna-myloxy) tetrahydro-2H-pyran-3,4,5-triyl triacetate(3)131 g (282 mmol),收率82%,mp.81~83 ℃。1H NMR (600 MHz, DMSO-d6, δ) 7.47 ~ 7.43 (m, 2H), 7.36 (dd, J = 8.4、7.0 Hz, 2H), 7.29 ~ 7.26 (m, 1H), 6.60 (dd, J = 16.0、1.7 Hz, 1H), 6.43 ~ 6.23 (m, 1H), 5.31 (t, J = 9.6 Hz, 1H), 4.98 ~ 4.89 (m, 2H), 4.84 (dd, J = 9.7、8.0 Hz, 1H), 4.40 (ddd, J = 13.3、5.5、1.6 Hz, 1H), 4.30 ~ 4.18 (m, 2H), 4.10 ~ 3.98 (m, 2H), 2.03 (d, J = 3.6 Hz, 6H), 2.00 (s, 3H), 1.95 (s, 3H)。13C NMR (151 MHz, DMSO-d6, δ) 170.51, 170.01, 169.74, 169.53, 136.71, 132.28, 129.11 (2 C), 128.24, 126.84 (2 C), 125.89, 99.18, 72.59, 71.51, 71.08, 69.57, 68.72, 62.23, 20.94, 20.88, 20.83, 20.73。ESI-MS C23H32NO10 [M+NH4]+ m/z=482.2030。将化合物(3)131 g (282 mmol)溶于1 L无水甲醇中,加入甲醇钠5 g,回流1 h,TLC检测反应完毕后,加入醋酸淬灭,然后蒸干甲醇,二氯甲烷∶甲醇-6∶1硅胶柱层析纯化,得化合物(4)78.4 g,收率95.2%。1H-NMR (300 MHz, DMSO-d6, δ) 7.43 (d, J = 7.2 Hz, 2H), 7.32 (t, J = 7.4 Hz, 2H), 7.23 (t, J = 7.2 Hz, 1H), 6.66 (d, J = 16.0 Hz, 1H), 6.35 (dt, J = 16.0, 5.7 Hz, 1H), 4.49 ~4.34 (m, 1H), 4.28 ~ 4.13 (m, 2H), 3.68 (d, J = 10.6 Hz, 1H), 3.45 (dd, J = 11.7, 5.3 Hz, 1H), 3.17 ~ 2.93 (m, 4H)。13C NMR (75 MHz, DMSO-d6) δ 136.97, 131.70, 129.09 (2 C), 128.05, 126.82, 126.77 (2 C), 102.61, 77.39, 77.22, 73.98, 70.57, 69.00, 61.58。ESI-MS C16H21O8[M+COOH]- m/z=341.1253。
-
将化合物(4)57 g(200 mmol)溶于吡啶中,冷却到0 ℃,然后分批加入叔丁基二苯基氯硅烷(TBDPSCl)52 g(200 mmol),加毕后升至室温,搅拌过夜,TLC检测,反应完毕,蒸干,硅胶柱层析得化合物(5)96.1 g,收率93.2%。1H NMR (600 MHz, DMSO-d6, δ) 7.76 ~ 7.67 (m, 4H), 7.49 ~7.39 (m, 9H), 7.33 (t, J = 7.6 Hz, 2H), 7.25 (t, J = 7.3 Hz, 1H), 6.66 (d, J = 16.0 Hz, 1H), 6.40 (dt, J = 16.0, 5.8 Hz, 1H), 4.53 ~ 4.41 (m, 1H), 4.31 (d, J = 7.8 Hz, 1H), 4.26 (dd, J = 13.7, 5.8 Hz, 1H), 4.02 ~ 3.97 (m, 1H), 3.80 (dd, J = 11.0, 6.0 Hz, 1H), 3.36 ~ 3.27 (m, 2H), 3.24 ~ 3.16 (m, 2H), 3.08 (t, J = 8.2 Hz, 1H), 1.02 (s, 9H)。13C NMR (151 MHz, DMSO-d6, δ) 136.88, 135.64 (2 C), 135.59 (2 C), 133.87, 133.76, 131.82, 130.21, 130.18, 129.09 (3 C), 128.23 (3 C), 128.08, 126.75 (3 C), 102.44, 77.22, 77.11, 73.93, 70.26, 68.75, 64.24, 27.06 (3 C), 19.40。ESI-MS C31H42NO6Si [M+NH4] +m/z=552.2781。
-
将化合物(5)96.1 g(180 mmol)加入300 ml醋酐,3 g二甲基氨基吡啶(DMAP),室温反应6~8 h。TLC检测反应完毕后,蒸干溶剂,加入乙酸乙酯溶解,加入1 mol/L盐酸洗涤,饱和碳酸氢钠洗涤,然后氯化钠洗涤,快速硅胶柱层析,得化合物(6)105 g,收率89%。1H NMR (600 MHz, DMSO-d6, δ) 7.70 (d, J = 6.7 Hz, 2H), 7.64 (d, J = 6.6 Hz, 2H), 7.51 ~ 7.40 (m, 9H), 7.33 (t, J = 7.6 Hz, 2H), 7.26 (t, J = 7.3 Hz, 1H), 6.59 (d, J = 16.0 Hz, 1H), 6.35 (dt, J = 16.0、5.8 Hz, 1H), 5.30 (t, J = 9.6 Hz, 1H), 5.16 (t, J = 9.7 Hz, 1H), 4.90 (d, J = 8.0 Hz, 1H), 4.88 ~ 4.79 (m, 1H), 4.44 (dd, J = 13.4、5.3 Hz, 1H), 4.27 (dd, J = 13.4, 6.2 Hz, 1H), 3.88 (dd, J = 12.5、2.8 Hz, 1H), 3.75 (s, 1H), 2.04 (s, 3H), 1.96 (d, J = 1.7 Hz, 6H), 1.00 (s, 9H)。13C NMR (600 MHz, DMSO-d6, δ) 170.09, 169.58, 169.44, 136.68, 135.69 (2 C), 135.59 (2 C), 133.32, 133.06, 132.15, 130.34, 129.09 (2 C), 128.31 (6 C), 126.83 (2 C), 125.97, 99.08, 73.58, 73.05, 71.61, 69.18, 68.41, 62.46, 26.92 (3 C), 20.95, 20.86 (2 C), 19.32。ESI-MS C37H48NO9Si [M+NH4] +m/z=678.3135。
-
将化合物(6)66 g(100 mmol)溶于300 ml四氢呋喃中,搅拌溶解后,0 ℃下加入吡啶40 ml、48%氢氟酸(30 ml),然后室温搅拌,TLC跟踪监测,反应完毕后,加入1 L乙酸乙酯,饱和碳酸氢钠洗涤,1 mol/L盐酸洗涤,无水硫酸钠干燥,硅胶柱层析纯化,得化合物(7)35.8 g,收率84%。1H NMR (600 MHz, DMSO-d6, δ) 7.45 (d, J = 7.5 Hz, 2H), 7.35 (t, J = 7.6 Hz, 2H), 7.27 (t, J = 7.3 Hz, 1H), 6.61 (d, J = 16.0 Hz, 1H), 6.34 (dt, J = 16.0, 5.7 Hz, 1H), 5.42 ~ 5.14 (m, 1H), 4.94 (t, J = 9.7 Hz, 1H), 4.88 ~ 4.80 (m, 2H), 4.45 (dd, J = 13.5, 4.9 Hz, 1H), 4.27 (dd, J = 13.5, 6.0 Hz, 1H), 3.73 (dd, J = 9.7、5.2、2.1 Hz, 1H), 3.56 (dd, J = 12.0、1.9 Hz, 1H), 3.46 (dd, J = 12.1、5.3 Hz, 1H), 2.02 (s, 3H), 1.99 (s, 3H), 1.95 (s, 3H). 13C NMR (151 MHz, DMSO-d6, δ) 170.08, 169.70, 169.55, 136.74, 132.12, 129.10 (2 C), 128.20, 126.83 (2 C), 125.97, 99.22, 74.22, 73.13, 71.69, 69.39, 69.10, 60.52, 20.94 (2 C), 20.81。ESI-MS C21H30NO9 [M+NH4]+ m/z=440.1922。
-
将1-羟基-2,3,4-三乙酰阿拉伯吡喃糖276 g(1000 mmol)溶于800 ml无水二氯甲烷中,加入三氯乙腈216 g(1500 mmol),搅拌均匀,冷却至0 ℃,滴加二氮杂二环(DBU)18 g(120 mmol),升至室温,反应3 h,TLC检测反应完毕,蒸干,石油醚:乙酸乙酯(2:1)快速硅胶柱层析,得无色油状物化合物(8)322 g,收率76%。1H NMR (600 MHz, DMSO-d6, δ) 9.88 (s, 1H), 6.45 (d, J = 3.7 Hz, 1H), 5.42 ~ 5.38 (m, 1H), 5.30 (dd, J = 10.7、3.4 Hz, 1H), 5.20 (dd, J = 10.8、3.5 Hz, 1H), 4.10 (d, J = 13.6 Hz, 1H), 3.89 (dd, J = 13.4、2.0 Hz, 1H), 2.14 (s, 3H), 2.01 (s, 3H), 1.99 (s, 3H). 13C NMR (151 MHz, DMSO, δ) 170.27, 170.24, 170.05, 158.69, 93.46 (2 C), 68.34, 67.26, 66.99, 63.06, 21.07, 20.89, 20.71。ESI-MS C13H20Cl3N2O8 [M+NH4]+ m/z=443.9826。
-
将化合物(7)42 g(100 mmol)、化合物(8)63 g(150 mmol)溶于500 ml无水二氯甲烷中,加入4Å分子筛100 g,氮气保护,室温下搅拌30 min,然后冷却到0 ℃,滴加三甲基硅三氟甲磺酸酯(TMSOTf)2.2 g(10 mmol),然后升至室温反应,TLC跟踪监测。反应完毕后,加入少量三乙胺淬灭反应,1 mol/L盐酸洗涤,饱和碳酸氢钠洗涤,无水硫酸钠干燥,蒸干,得乙酰络塞维,无须纯化,直接进行下一步反应。将乙酰络塞维粗品62 g(90 mmol)溶于无水甲醇中,溶解,再加甲醇钠5 g,回流3 h,反应完毕后,加入醋酸淬灭,然后蒸干甲醇,C-18液相分离,得白色固体络塞维32 g,两步收率75.7%,mp.171~173 ℃。1H NMR (600 MHz, DMSO-d6, δ) 7.43 (d, J = 7.8 Hz, 2H), 7.32 (t, J = 7.6 Hz, 2H), 7.27 ~ 7.19 (m, 1H), 6.66 (dd, J = 16.0、3.8 Hz, 1H), 6.44 ~ 6.26 (m, 1H), 5.08 (t, J = 4.4 Hz, 1H), 5.00 ~ 4.92 (m, 2H), 4.82 (dd, J = 13.2、3.1 Hz, 1H), 4.53 (d, J = 5.7 Hz, 1H), 4.47 (d, J = 4.5 Hz, 1H), 4.40 (ddd, J = 13.2、5.3、1.7 Hz, 1H), 4.21 (dd, J = 11.1、4.9 Hz, 3H), 3.93 (dd, J = 11.3、1.7 Hz, 1H), 3.84 ~ 3.69 (m, 1H), 3.69 ~ 3.59 (m, 2H), 3.57 ~ 3.27 (m, 8H), 3.14 (td, J = 8.9、4.8 Hz, 1H), 3.08 ~ 2.96 (m, 2H). 13C NMR (151 MHz, DMSO-d6, δ) 136.98, 131.98, 129.08 (2 C), 128.05, 126.80 (2 C), 126.70, 103.98, 102.37, 77.09, 76.15, 73.89, 73.01, 71.02, 70.66, 68.93, 68.61, 67.79, 65.31。ESI-MS C20H28O10Na [M+Na]+ m/z=451.1580。
-
本研究所用的合成方法以β-D-五乙酰基葡萄糖和阿拉伯吡喃糖为起始原料,合成1-O-三氯亚胺酯-2,3,4,6四乙酰葡萄糖、1-O-三氯亚胺酯-2,3,4,三乙酰阿拉伯糖为高活性糖基化供体,合成络思为中间体,经选择性保护和脱保护后,以三氟甲磺酸三甲基硅脂为糖苷化反应催化剂合成络塞维,此方法较三氟化硼乙醚糖基化收率更高[5],比碳酸银、三氟甲磺酸银[6]催化溴代四乙酰葡萄糖的方法更为简单,无需避光操作,而且原料便宜,成本更低,合成过程中无高危险性反应。三氯亚胺酯为活化基团的供体,较端位乙酰基糖为活化基团供体的糖基化反应收率更高。在葡萄糖6位-OH的选择性保护上,叔丁基二苯基硅基[7, 8]以足够的空间位阻,具有高效的选择性,稳定性较好,较-Li[4]用三苯甲基保护更有优势,而且后期选择性脱除简单,极大简化了合成过程,提高了合成中间体的反应收率。本方法合成络塞维工艺简单,合成路径短,生产成本低,原料便宜,安全性高,污染少,收率高,适合于络塞维的规模性放大生产。
Synthesis of the active ingredient rosavin of Rhodiola rosea
-
摘要:
目的 研究红景天活性成分络塞维(rosavin)的化学全合成方法。 方法 以β-D-五乙酰葡萄糖、1-羟基-2,3,4-三乙酰阿拉伯吡喃糖、肉桂醇为起始原料,经1-位选择性脱乙酰基、糖苷化反应、葡萄糖6-OH选择性保护及脱保护等8步反应制备目标化合物。 结果 以高收率成功获得目标产物络塞维,结构经ESI-MS和1H-NMR、13C-NMR确证。首次报道以叔丁基二苯基氯硅烷高选择性、高收率保护6-OH的技术途径。 结论 该合成路线具有操作简单,收率高,安全性好等优点。 Abstract:Objective To establish the chemical synthesis of the active ingredient rosavin of Rhodiola rosea. Methods β-D-pentaacetylglucose, 1-hydroxy-2,3,4-triacetylarabinose and cinnamyl alcohol were used as starting materials. The target compound was prepared by 1-position selective of β-D-pentaacetylglucose deacetylation, glycosylation reaction, glucose 6-OH selective protection and deprotection and other 8-step reactions. Results The target product, rosavage, was successfully obtained with high yield. The structure was confirmed by ESI-MS, 1H-NMR and 13C-NMR. The protection of 6-OH with high selectivity and high yield of tert-butyldiphenyl chlorosilane played a vital role in the synthesis process,. Conclusion The synthetic route has the advantages of simple operation, high yield, and good safety. -
Key words:
- Rhodiola rosea /
- rosavin /
- total synthesis /
- selective protection
-
[1] 侯奋争, 姚桂彬, 徐伟, 等. 高山红景天首次分离的化合物(Ⅰ)[J]. 中国现代中药, 2009, 11(4):18-20. [2] 于冬建. 红景天药理学研究及临床作用新进展[J]. 中医临床研究, 2020, 12(18):136-138. [3] 刘素欣, 张露, 崔承弼. 红景天提取物的降血糖作用研究[J]. 延边大学农学学报, 2020, 42(2):21-26. [4] LI X D, KANG S T, LI G Y, et al. Synthesis of some phenylpropanoid glycosides (PPGs) and their acetylcholinesterase/xanthine oxidase inhibitory activities[J]. Molecules,2011,16(5):3580. doi: 10.3390/molecules16053580 [5] ABEYRATHNE A R N M, PERERA A D L C, KARUNARATNE D N. Liquid crystal behaviour of three novel glycosides[J]. J Natl Sci Found Sri Lanka,2012,40(2):115. doi: 10.4038/jnsfsr.v40i2.4439 [6] ZHANG Q, PENG X R, GRILLEY M, et al. Using fluorogenic probes for the investigation of selective biomass degradation by fungi[J]. Green Chem,2015,17(3):1918-1925. doi: 10.1039/C4GC01659A [7] CMOCH P, PAKULSKI Z. Comparative investigations on the regioselective mannosylation of 2, 3, 4-triols of mannose[J]. Tetrahedron: Asymmetry,2008,19(12):1494-1503. doi: 10.1016/j.tetasy.2008.05.032 [8] DE SOUZA A, HALKES K, MEELDIJK J, et al. Synthesis of gold glyconanoparticles: possible probes for the exploration of carbohydrate-mediated self-recognition of marine sponge cells[J]. Eur J Org Chem,2004,2004(21):4323-4339. doi: 10.1002/ejoc.200400255 -

 点击查看大图
点击查看大图
图(1)
计量
- 文章访问数: 9268
- HTML全文浏览量: 3427
- PDF下载量: 51
- 被引次数: 0
