

 下载:
下载:



-
试验用药品指用于临床试验的试验药物、对照药品[1-3]。据国家药品监督管理局食品药品审核查验中心于2017年发布数据显示,2015年7月~2017年6月对313个药品注册现场核查报告中所发现的缺陷进行分类,其中与试验用药品相关的缺陷占11.6%,在所有缺陷中排名第3位[4]。试验用药品管理作为临床试验全流程管理中的关键步骤,直接影响到临床试验的质量[5-6]。因此,摸清我国试验用药品管理的研究现状与存在问题,探索解决或改进方案,有助于切实提高药物临床试验质量[7-8]。据此,笔者采用文献计量学的相关方法,检索中国知网、万方医学网、维普网等中文数据库中公开发表的试验用药品管理相关文献,挖掘该领域目前的研究现状与发展趋势,为相关工作者优化试验用药品管理模式、提高临床试验质量提供一定的参考。
-
在中国知网、维普网、万方医学网等3个中文数据库的“高级检索”模块中,在“题名”处分别输入“试验”、“药品”、“管理”等检索词,之间以“和”或“and”进行连接,检索时段为建库日至2022年12月。记录收集检索文献的题名、所有作者姓名、所有作者单位、资助基金名称、中文摘要、所有中文关键词、发表年限和期刊名称。
-
①题名中含有所有检索词的文献;②以我国为研究背景的文献;③正式发表的文献(非网络首发文献)。
-
①在不同数据库中收录的同一篇文献,仅纳入一篇;②同一作者在不同载体上发表的同一篇文献(如期刊论文与会议论文),仅纳入一篇;③不同作者发表的高度雷同的文献;④以国外为研究背景的文献;⑤消息、报道、通知、征文启事、科普类非正规文献。
-
提取纳入文献的题名、作者姓名、作者单位、资助基金、中文摘要及关键词,以refworks格式将以上信息导出,由专人进行核对。导出的信息经中国知网的文献可视化分析模块、CiteSpace等进行聚类分析,绘制知识图谱与柱状图。
-
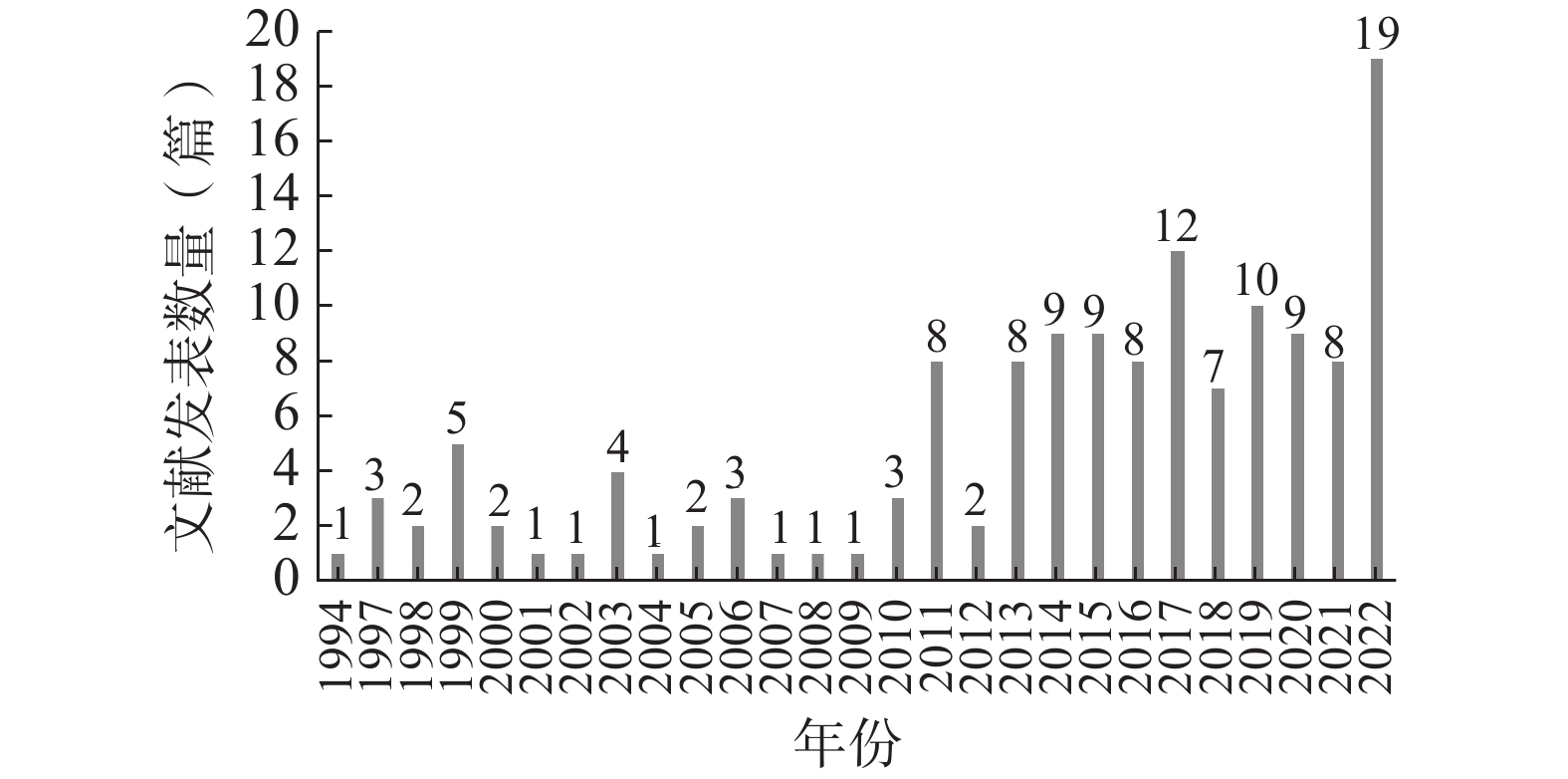
以我国试验用药品管理为主要内容的文献最早发表于1994年,截至2022年12月共计发表140篇,其中发表文献数量最多的年份为2022年(19篇)。自1994年至2022年,该领域的年度文献发表数量不断波动,但总体呈上升趋势(图1)。
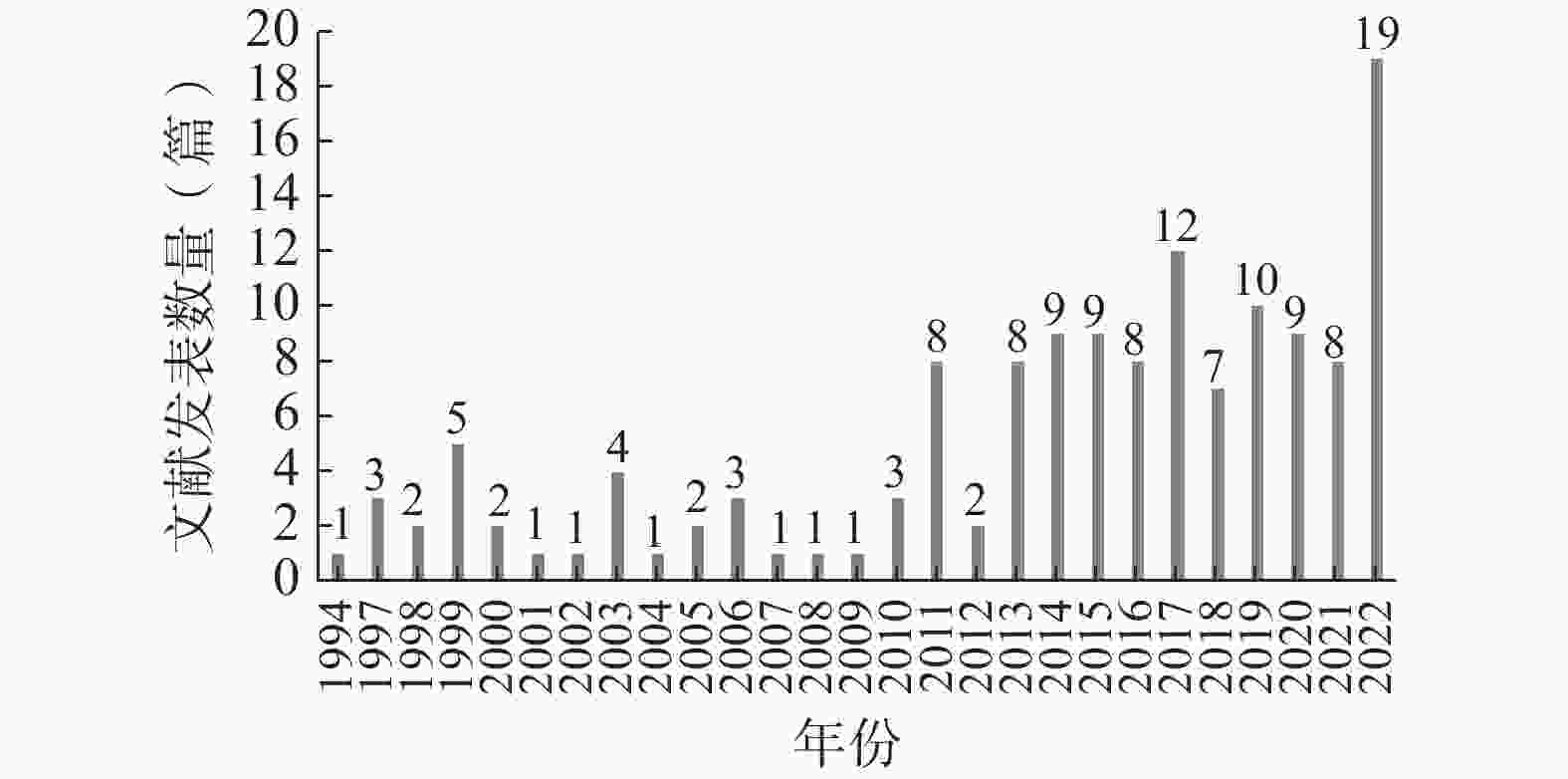
图 1 年度文献发表数量统计图
-
提取所有纳入文献的关键词并共现统计,将同一篇文献中的关键词绘制连接线,最终绘制网络图谱。按关键词共现频次由高至低排序,前10个关键词依次为:受试者(24次)、研究者(15次)、药品(14次)、伦理委员会(13次)、药品临床试验(13次)、临床研究(11次)、试验方案(10次)、知情同意书(8次)、管理规范(8次)、指导原则(8次)、试验(8次),具体见图2。
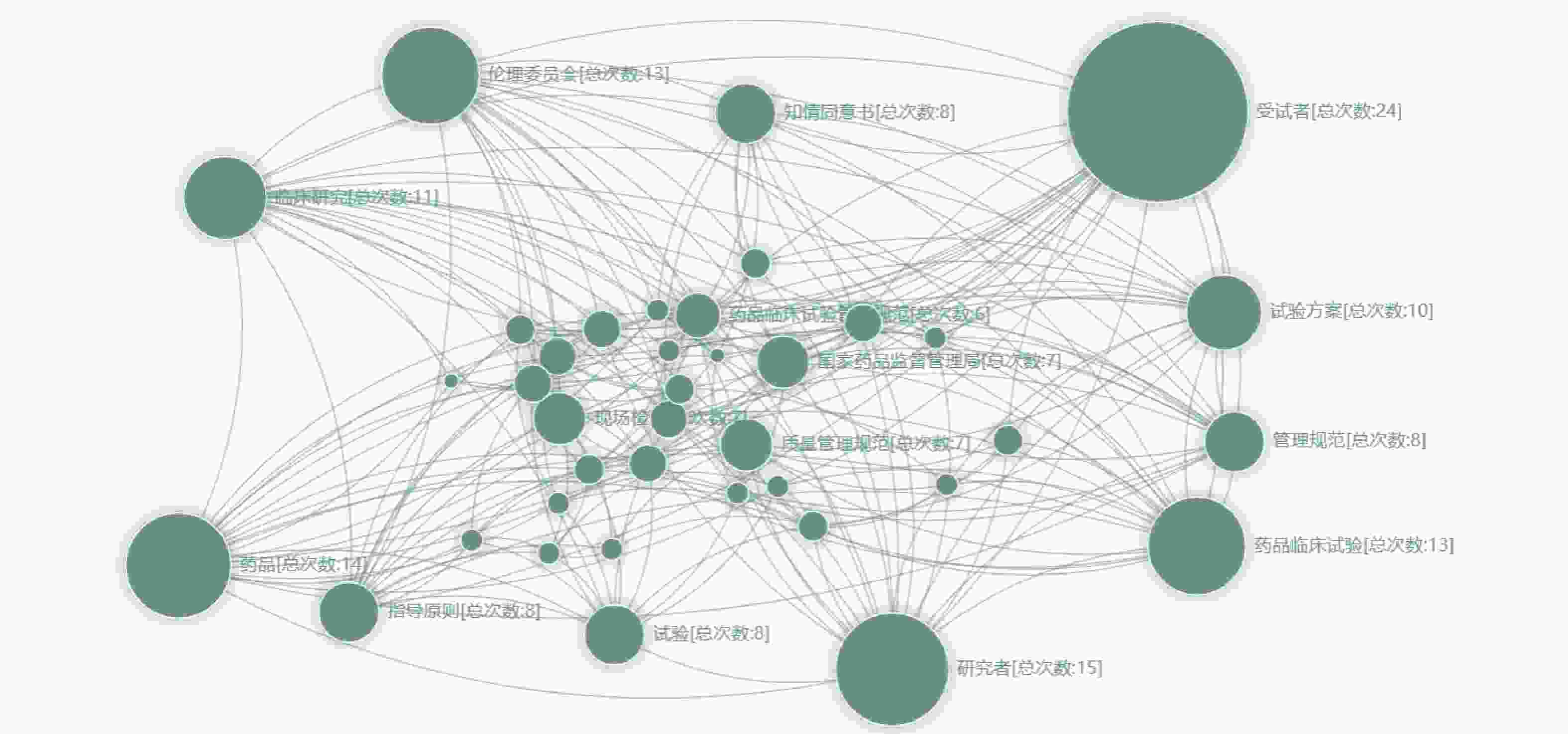
图 2 关键词共现网络图谱
-
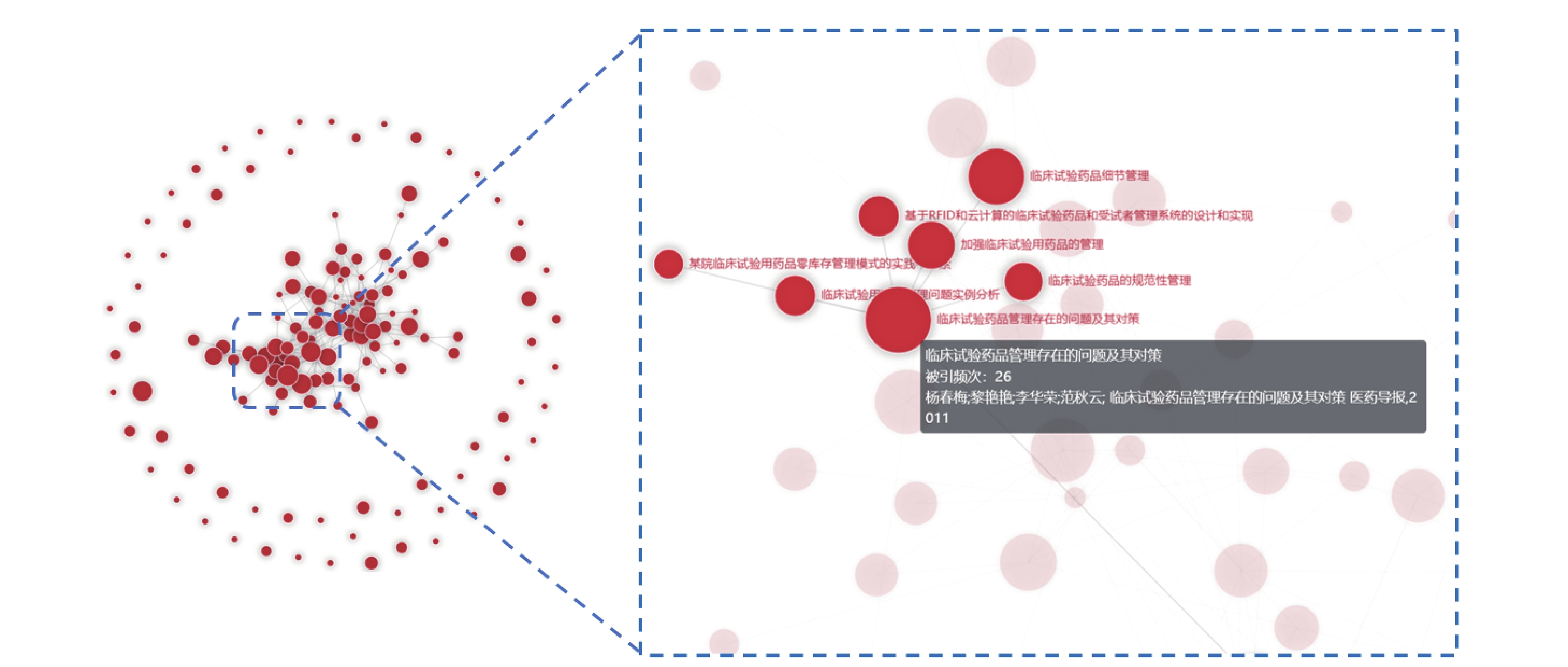
图3为文献互引网络图谱,被引频次最高的文献是华中科技大学同济医学院附属荆州医院药物临床试验机构的杨春梅等[9]在2011年发表于《医药导报》的《临床试验药品管理存在的问题及其对策》,被引频次为26次,占比18.57%。
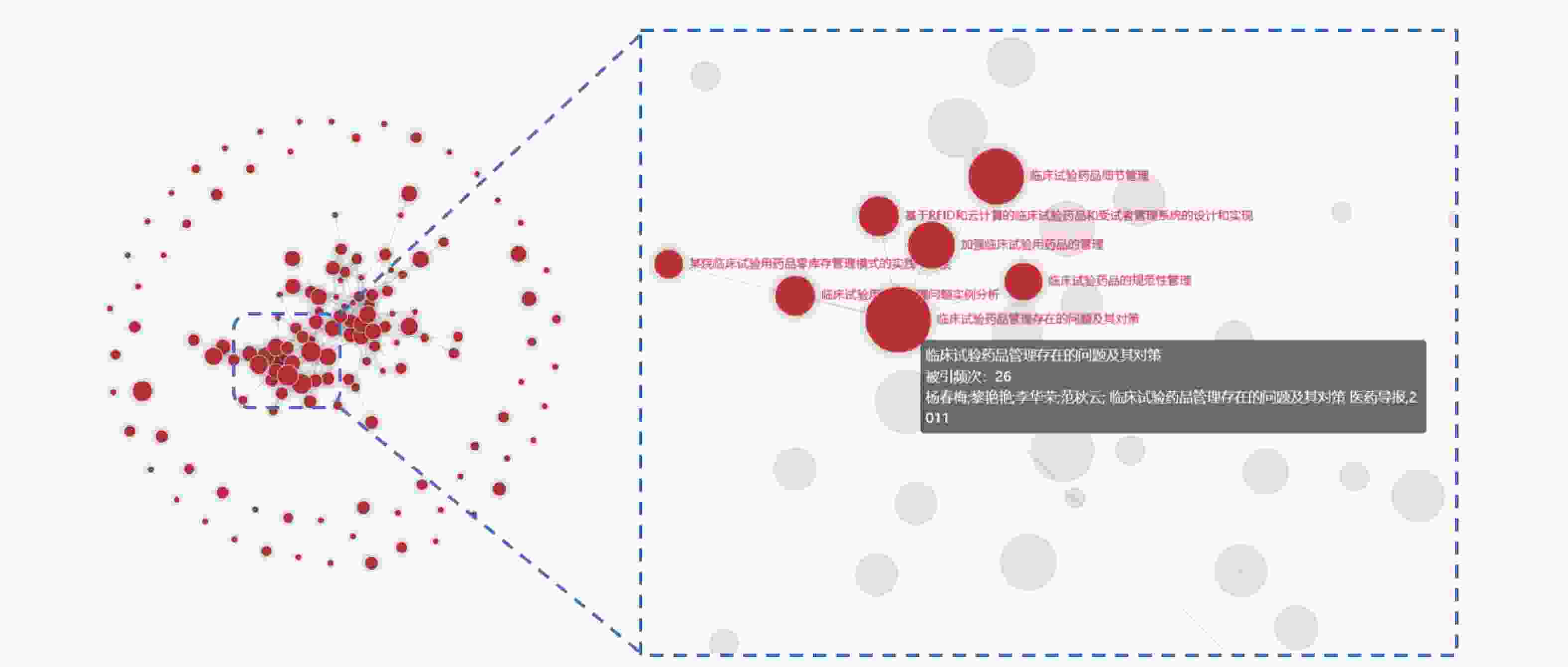
图 3 文献互引网络图谱
-
提取所有纳入文献的作者姓名并共现统计,同一作者的多篇文献进行合并(发文数量与点的大小呈正相关),并将同一篇文献中的各作者间绘制连接线,最终绘制作者合作网络图谱。如图4所示,我国试验用药品管理领域的作者间合作较为松散,团队间合作交流较少,其中合作较为密切的为北京大学第三医院的杨丽团队与陈凤荣团队。

图 4 作者合作网络图谱
-
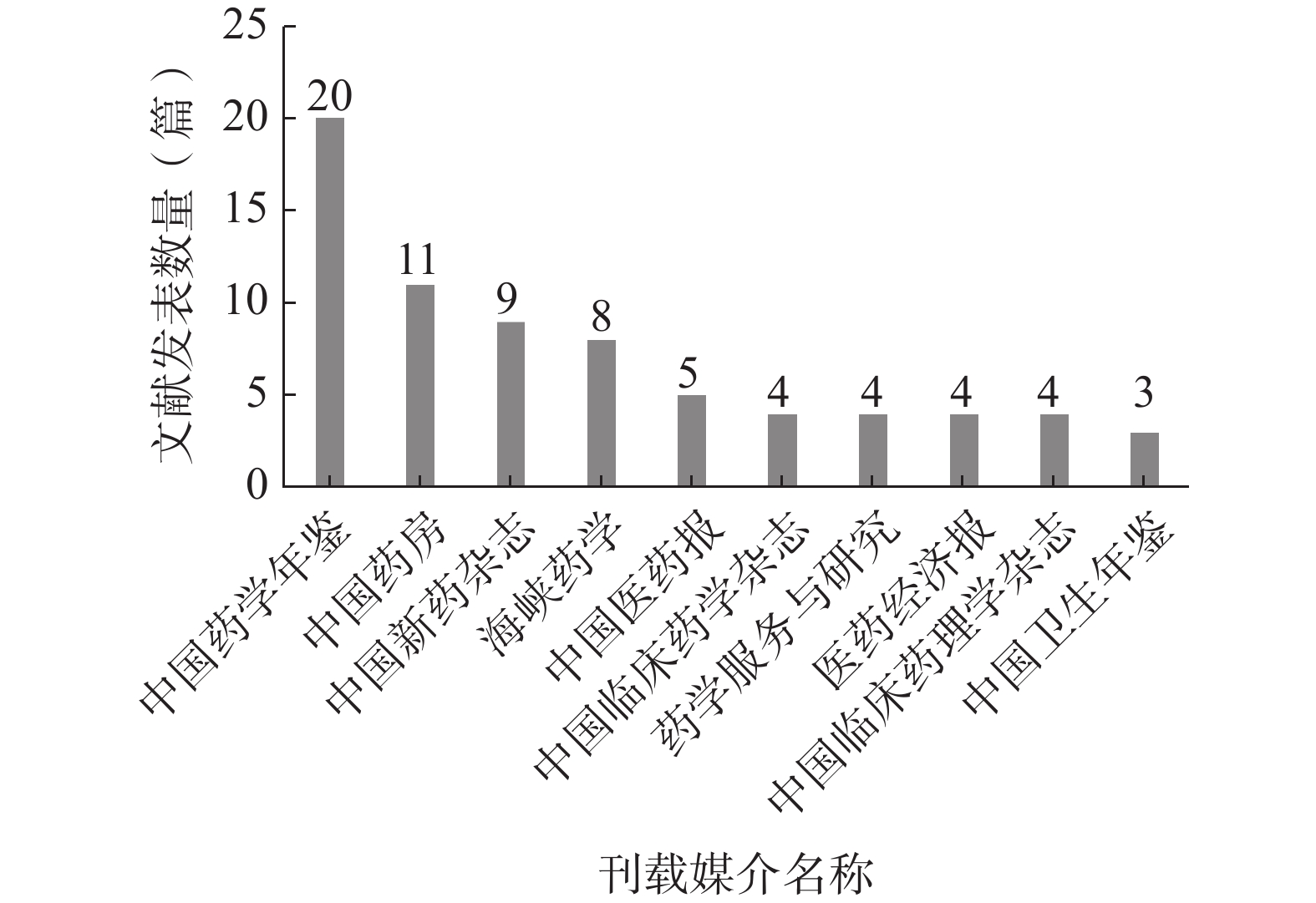
统计所有纳入文献的刊载媒介,按发文数量由高至低进行排序,发文数量最多的刊载媒介为《中国药学年鉴》,发文数量前10名的刊载媒介见图5。
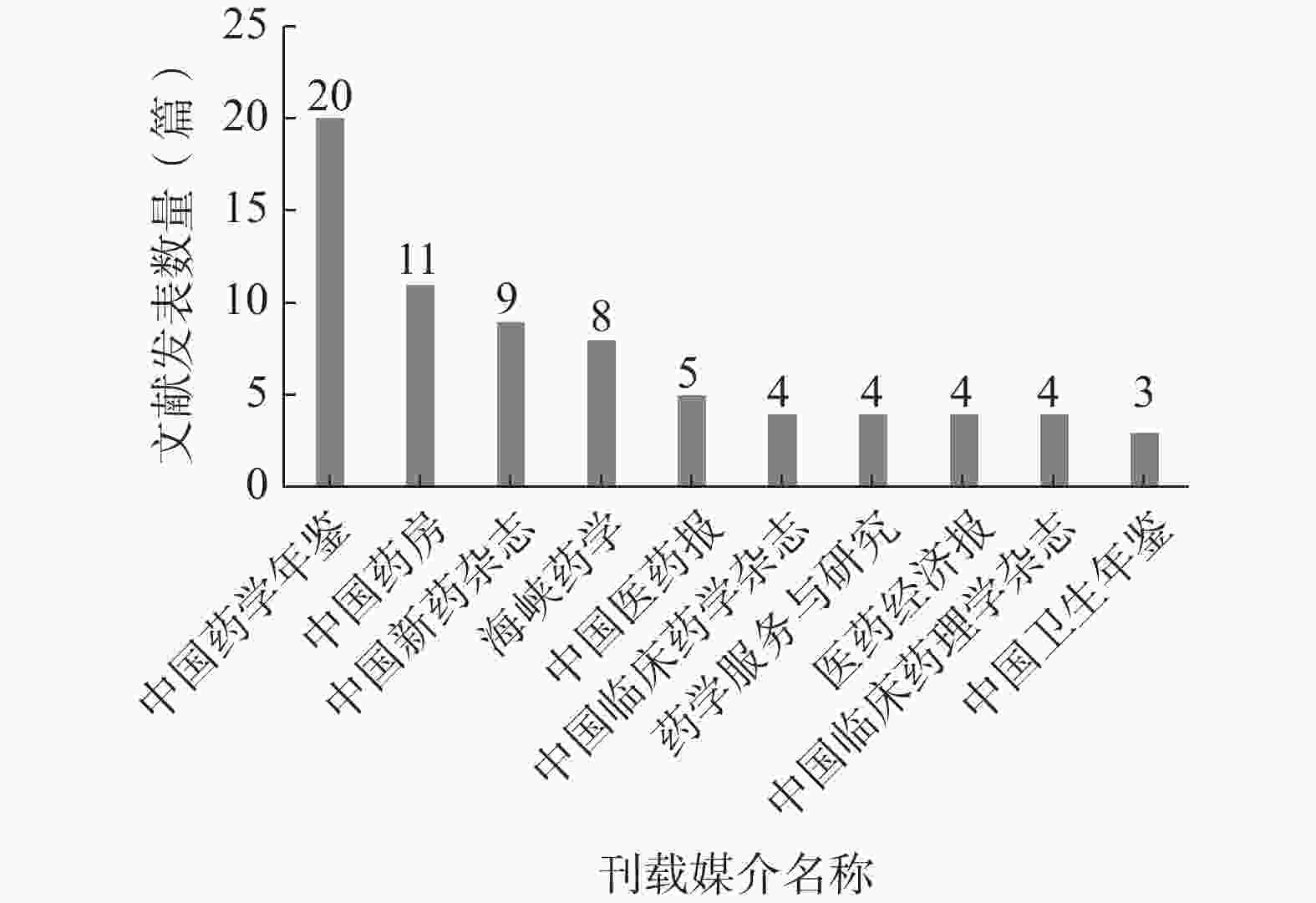
图 5 前10名刊载媒介名称
-
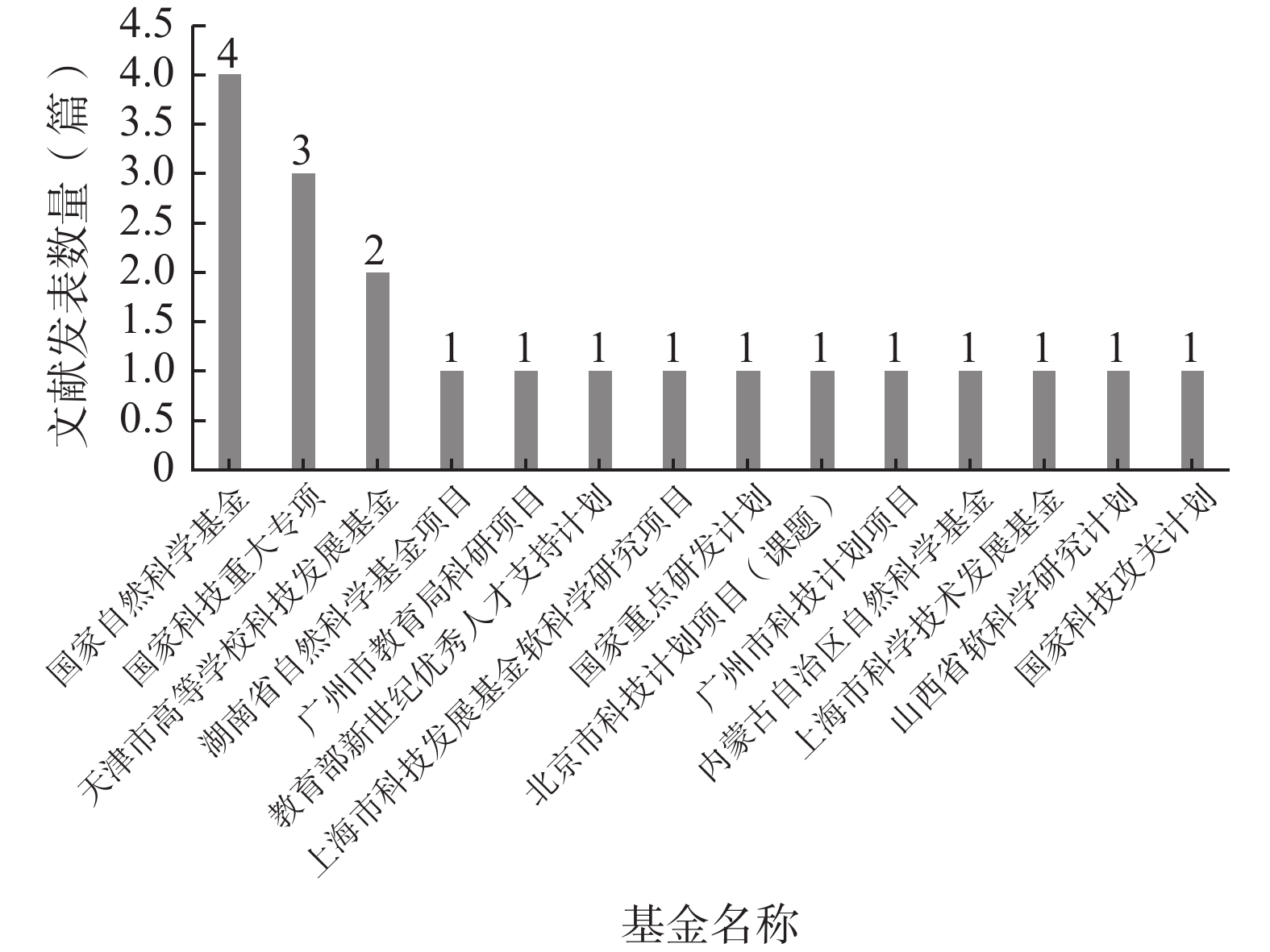
如图6所示,在140篇纳入文献中,仅有20篇文献得到了基金资助,资助率仅为14.29%,其中资助该研究领域最多的基金为国家自然科学基金(4篇)。
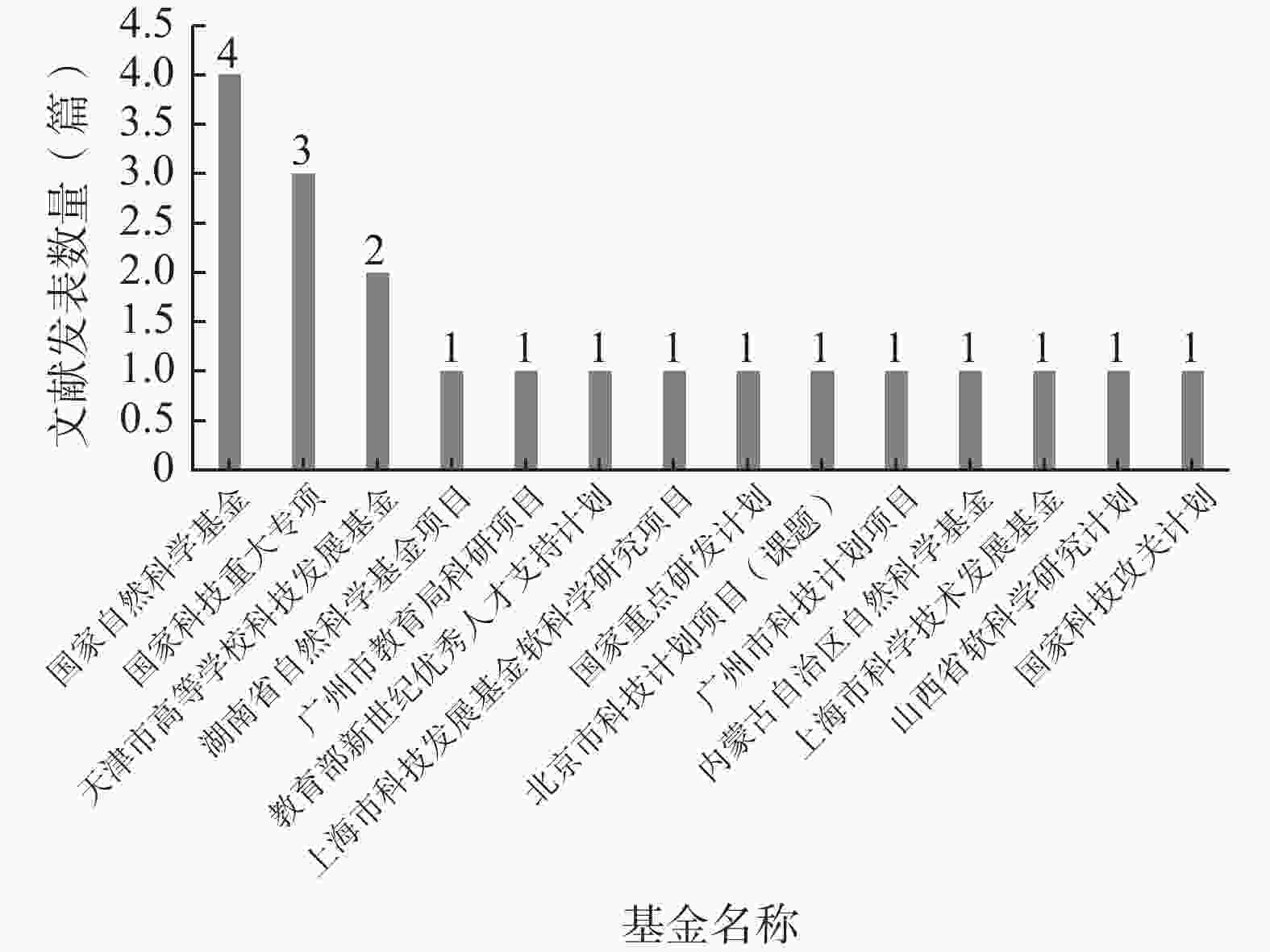
图 6 基金资助统计图
-
自1994年至2010年,我国试验用药品管理领域的发文数量较为低迷,且不断上下波动。自2011年始,发文量有显著提高,且于2022年达到峰值。整体上看,该领域的年度发文量呈上升趋势,但增势较缓。这一方面是由于我国临床试验的参与人数总体较少,尤其是医疗机构内参与临床试验的人数更少,导致临床试验类文献数量总体较少;另一方面是由于我国参与临床试验的人员中,对试验用药品管理的重视不够、关注不足,导致该领域的年度发文量较为低迷。
-
对图2中权重最大的10个关键词进行分析,剔除与检索词同义的关键词(药品、药品临床试验、临床研究、试验)后,得出试验用药品管理领域较为重要的关键词为:受试者、研究者、伦理委员会、试验方案、知情同意书、管理规范、指导原则。
其中,权重最大的关键词为“受试者”,这表明在试验用药品管理领域,大部分作者的首要关注点为受试者。这与《药物临床试验质量管理规范》(GCP)的基本理念相一致,保证受试者的安全是临床试验的第一要义,而对试验用药品的规范管理,确保药品的质量合格,则是保证受试者安全的第一步[10-11]。此外,受试者也是试验用药品管理最直观的评价者。现如今,医疗机构均贯彻“以患者为中心”的服务理念[12-13],故临床试验也应“以受试者为中心”,因此对受试者进行充分的用药指导显得尤为重要。受试者对药品的用法用量及疗程是否清楚、是否了解试验结束后应退回哪些物品、对研究医师与药品管理员的服务是否满意等,均是管理质量的重要考查指标。基于以上原因,“受试者”成为图谱中权重最大的关键词。
与“受试者”相对的关键词是“研究者”,研究者是试验用药品管理的具体实施者,主要包括研究医师、研究护师、药品管理员3种角色[14]。一方面,研究者保证试验用药品管理质量的先决条件则是对“试验方案”(关键词)的充分理解,若研究者对试验方案的理解有偏差,导致给受试者错用、误用试验药品,将严重危害受试者的安全,亦会影响整体的试验结果[15-16]。另一方面,试验方案应是科学、严谨、合理的,但在实施过程中,由于各医疗机构软/硬件、人员构成、运行制度等方面的差异,会出现无法完全按照试验方案执行项目的情况,若遇到此种情况,研究者应及时与申办方沟通、协调,按实际情况修订试验方案,并提交伦理委员会审理,待伦理委员会通过后再进行具体实施[17-18]。由此,又引出了“伦理委员会”这一关键词。伦理委员会的主要职责是保证临床试验的安全性、科学性、合理性,虽然其不直接参与试验用药品的管理过程,但药品信息变更(如药品新增批号、药品有效期延长等)均需在伦理委员会进行备案,若受试者在使用试验药物后出现严重不良事件或可疑且非预期严重不良反应也应及时向伦理委员会报告[19-20]。
而与“伦理委员会”关系最为密切的关键词则是“知情同意书”。知情同意书是伦理委员会对临床试验进行立项审核的重要内容,也是受试者全面了解临床试验及药品信息的主要渠道,因此在知情同意书中应用通俗易懂的文字详细叙述试验药品的背景信息、用法用量、疗程以及可能发生的不良反应等,努力做到受试者的充分知情。
在图2中“受试者-研究者-试验方案-伦理委员会-知情同意书”之间存在着紧密的逻辑关系,同时也是临床试验过程中较为关注的领域,因此共现权重较大且逻辑线繁密。而保证以上5个关键词合法、合规的法律基础则是GCP及各种“指导原则”,这导致“管理规范”与“指导原则”两个关键词在图谱中的共现权重也较大。其中,GCP是药物临床试验全过程的质量标准,临床试验的任何步骤都应遵循该规范 [21-22]。而指导原则泛指的内容较多,国家药品监督管理局药品审评中心针对不同类型的药物、不同类型的临床试验制定了相应的指导原则[23-25]。截止2023年11月22日,已有429个指导原则。
-
在纳入文献中,被引频次最高的文献为《临床试验药品管理存在的问题及其对策》,但较为陈旧(发表于2011年),且在2020年我国颁布新版GCP后,该文献的部分内容已与现行规章有所出入,由此可见试验用药品管理领域的发展较为缓慢、滞后,需尽快得到临床试验从业人员的重视。
-
在试验用药品管理研究领域,影响权重较大的作者主要来自三甲医院,这是由于我国绝大多数临床试验是在三甲医院开展的,且三甲医院的品牌效应也吸纳了较多医疗人才,因此临床试验的从业人员较多。但是,图谱显示该领域的作者间的合作较少,合作网络较为简单,也未出现较有影响力的领军人物,这可能是由于各基金评审部门在该领域缺乏导向性,故无法促成各研究者进行合作,从而导致该领域的发展较为迟缓。
-
对发文数量前10名的刊载媒介按刊载媒介的种类分类,其中期刊类媒介6个、年鉴类媒介2个、报刊类媒介2个;而在6个期刊类媒介中,3个期刊隶属北大中文核心期刊,1个期刊隶属中国科技核心期刊(统计源),2个期刊为“双非”期刊。该结果表明试验用药品管理研究领域的文献质量整体偏低,发展水平较为落后。
-
在纳入文献中,基金资助率仅为14.29%,说明我国的各基金评审部门对该领域的重视不足,这也是该领域文献的质量较低的原因之一。因此,建议相关部门能够增强对试验用药品管理的资助力度,通过设立专项基金的方式吸引更多的医药工作者开展该领域的研究工作,从而推动该领域的发展。
-
现阶段我国试验用药品管理领域关注的重点为“受试者-研究者-试验方案-伦理委员会-知情同意书”这一逻辑线,其中最为关注的是受试者的用药安全与用药规范,这直接决定了对试验药物的安全性与有效性评价,也直接决定了试验药物能否上市。据笔者调研,以上逻辑线也是国际上试验用药品管理领域比较关注的重点内容。然而,我国现阶段在此领域的文献数量偏少且质量较低,作者间的合作也不紧密、交流较少,各基金项目对此领域的资助力度也有所欠缺,以上因素导致该领域的整体发展水平较低、发展速度较为缓慢。因此,呼吁相关医药工作者对以上问题予以重视,探索解决或改进方案,促进试验用药品管理领域的发展与完善,从而有效保障受试者的人身安全,切实提高我国药物临床试验的质量。
Data mining of current research status of clinical trial drug management in China by bibliometrics
-
摘要:
目的 基于文献计量学方法对我国试验用药品管理领域的文献进行数据挖掘,明确该领域的发展现状与研究热点。 方法 在中国知网等3家中文文献数据库中,以“试验”、“药品”、“管理”为检索词,用“and”或“和”连接,从文献中提取文献题名、作者姓名、作者单位名称、中文摘要、中文关键词、发表年限、期刊名称等内容,根据中国知网文献可视化分析系统、CiteSpace等软件进行聚类分析并绘制网状知识图谱。 结果 我国试验用药品管理领域的文献最早发表于1994年,至2022年共计发表文献140篇,其中有20篇文献得到了相关基金资助,关键词共现频次最高的是“受试者”,刊载文献最多的媒介为《中国药学年鉴》。 结论 现阶段我国试验用药品管理领域的文献数量较少且质量较低,作者间的合作也不紧密、交流较少,各基金项目对此领域的资助力度也有所欠缺,以上因素导致该领域的整体发展水平较低、发展速度较为缓慢。 Abstract:Objective To clarify the current development status and research hotspots in the field of experimental drug management in China through data mining by bibliometric. Methods Key words such as “experiment”, “drug”, and “management” were used the search the Chinese literature in China National Knowledge Infrastructure (CNKI). The title, author name, author affiliation, Chinese abstract, Chinese keywords, publication period, journal name, and other content of the literature were extracted from the literature. Cluster analysis was performed by CNKI literature visualization analysis system, CiteSpace and other software, and a network knowledge map was drawn. Results The literature in the field of experimental drug management in China was first published in 1994, and a total of 140 articles were published until 2022. Among them, 20 articles were supported by relevant funds, and the keyword co-occurrence frequency was highest among “subjects”. The most frequently published medium was the Chinese Pharmacological Yearbook. Conclusion At present, the quantity and quality of literature in the field of experimental drug management in China are relatively small, and the cooperation and communication among authors are not close. The funding from various fund projects in this field is also lacking. These factors lead to a lower overall development level and slower development speed in this field. -
Key words:
- clinical trials /
- medicines /
- management /
- bibliometrics /
- data mining
-
[1] 国家药品监督管理局, 国家卫生健康委员会. 关于发布药物临床试验质量管理规范的公告(2020年第57号)[EB/OL].(2020-04-26)[2024-03-15]. https://www.nmpa.gov.cn/xxgk/fgwj/xzhgfxwj/20200426162401243.html. [2] 国家药品监督管理局食品药品审核查验中心. 药物临床试验数据核查阶段性报告(2015年7月-2017年6月)[EB/OL].(2017-07-21)[2024-03-15]. https://www. cfdi.org.cn/resource/news/9137.html. [3] 国家药品监督管理局食品药品审核查验中心. 药品注册核查要点与判定原则(药物临床试验)(试行)(2021年第30号)[EB/OL].(2020-12-17)[2024-03-15]. https://www.cfdi.org.cn/resource/news/14199.html. [4] 国家药品监督管理局, 国家卫生健康委员会. 关于发布药物临床试验机构管理规定的公告(2019年第101号)[EB/OL].(2019-11-29)[2024-03-15]. https://www.nmpa.gov.cn/xxgk/fgwj/xzhgfxwj/20191129174401214.html. [5] 广东省药学会. 药物临床试验制度建设·广东共识(2020年版)[EB/OL].(2020-08-01)[2024-03-15]. http://www.sinopharmacy.com.cn/notification/2003.html. [6] 朱露莎, 季晓慧, 孔敏, 等. PDCA循环法用于试验用药品管理质量改进效果分析[J]. 中国药业, 2022, 31(15):16-20. doi: 10.3969/j.issn.1006-4931.2022.15.003 [7] 甘园, 张琴, 黄燕萍, 等. 新的药物临床试验机构开展药物临床试验工作的实践与思考[J]. 中国新药杂志, 2019, 28(22):2749-2753. [8] 于文惠, 梁雁, 赵侠, 等. 基于GCP药房的药物临床试验质量管理[J]. 中国新药杂志, 2019, 28(3):314-318. [9] 杨春梅, 黎艳艳, 李华荣, 等. 临床试验药品管理存在的问题及其对策[J]. 医药导报, 2011, 30(6):829-830. doi: 10.3870/yydb.2011.06.050 [10] 马丽平, 李语玲, 王铁军, 等. 药师在临床试验药物管理过程中的风险防控与思考[J]. 中南药学, 2021, 19(10):2217-2220. [11] 王彦荣, 李卫红, 郭鹏, 等. 我院临床试验药房的建设与管理[J]. 中国药业, 2021, 30(1):24-27. [12] 吴伟, 李劲彤. PDCA循环在临床试验质量控制中的应用[J]. 中国临床药理学杂志, 2020, 36(3):377-378. [13] 孟啸, 陈庆琳, 朱玉洁, 等. 药物临床试验医院信息系统互联互通实践研究[J]. 中国医院药学杂志, 2020, 40(5):565-569. [14] 蔡君龙, 周晶晶, 李晓晖, 等. 临床试验用药品信息系统管理专家共识[J]. 药物评价研究, 2021, 44(5):917-923. [15] 刘金永, 李子玥. 药物临床试验过程中试验用药品管理的实践体会[J]. 中国药物评价, 2020, 37(5):391-393. [16] 朱露莎, 季晓慧, 孔敏, 等. 试验用药品超温原因分析和处理对策[J]. 中医药管理杂志, 2021, 7(23):168-169. [17] 裴彤, 胡朝英, 胡晓, 等. 中美药物临床试验中的药品管理现状比较[J]. 中国药房, 2019, 30(3):294-298. [18] 唐铭婧, 梅和坤, 江学维, 等. 专职药师在临床试验用药品管理中的重要作用[J]. 中国新药杂志, 2017, 26(22):2710-2713. [19] 徐佳琦. 医院药物临床试验中心药房的标准化建设和规范化管理[J]. 中国现代应用药学, 2019, 5(23):2978-2982. [20] 王思颖, 汤萍, 熊艳, 等. 药物临床试验质量控制与对策分析[J]. 中国现代医药杂志, 2020, 22(8):80-82. [21] 孙雪, 宋浩静, 郭彩会, 等. 信息化系统对药物临床试验质量的影响[J]. 中南药学, 2019, 17(6):937-940. [22] 黄义昆, 毛晓丽, 李红梅, 等. 临床试验用药品的中心化管理[J]. 中国医药科学, 2021, 11(18):228-230. doi: 10.3969/j.issn.2095-0616.2021.18.059 [23] 袁婷婷, 余自成, 杜静, 等. 药师在药物临床试验中的角色与定位[J]. 中南药学, 2021, 19(3):553-556. [24] 周心娜, 王淑梅, 谢铮铮, 等. 临床试验用药物的质量控制[J]. 中国临床药理学杂志, 2019, 5(12):1306-1307. [25] 杨忠奇, 赖育健, 吴波林, 等. 药物临床试验 药物管理·广东共识(2014年)[J]. 今日药学, 2015, 1(2):75-76. -

 点击查看大图
点击查看大图
图(6)
计量
- 文章访问数: 13501
- HTML全文浏览量: 1843
- PDF下载量: 19
- 被引次数: 0
