

 下载:
下载:

-
曲马多是一种具有阿片受体激动剂性质的合成镇痛药,一般用于急、慢性疼痛,中、轻度癌症疼痛,骨折或各种术后疼痛、牙痛等[1],长期使用曲马多可导致成瘾。由于曲马多的缓释制剂、复方制剂在中度及以上疼痛的老年患者中应用范围和使用剂量不断扩大,曲马多引起的不良反应、药物相互作用等潜在不合理用药(PIM)案例报道呈现上升趋势[2]。2023年7月1日起,中国将曲马多复方制剂列入第二类精神药品目录。建立快速测量患者体液中曲马多的方法,有助于减少该类药物滥用和潜在不合理用药。
口服盐酸曲马多约60%的剂量由肾脏代谢,29%的剂量以原型经尿液排出体外[3]。目前检测体液中曲马多的方法一般是采用高效液相色谱-紫外检测法(HPLC-UV)和气相色谱-质谱联用法(GC-MS)[4-5],但这些方法灵敏度低,且相对耗时、费力,检测成本较高,很难在基层的临床药品检测中大规模推广。分子光谱技术由于其快速、无损、高效、简单的特点,如拉曼光谱技术,近几年在临床药品检测中逐渐被推广应用[6-9],如用于快速监测血清中卡马西平[8]以及尿液中氯氮平的监测[9]。2015年,Alharbi等[10]利用表面增强拉曼光谱(SERS)检测模拟人工尿液中曲马多的含量,其检测限(LOD)可以达到657.5 ng/ml,已经非常接近临床使用曲马多需要检测的药物浓度。
为了尽量减少尿液中成分对SERS信号影响,本研究提出用液液萃取(LLE)来改进基于SERS的尿液中待测成分检测[11-12]。LLE-SERS法测定患者服用曲马多后的真实尿液样品,能够快速确认患者是否服用曲马多以及监测尿药浓度,有助于确定曲马多在缓解老年患者疼痛时的个体化合理剂量,最大程度降低因曲马多药物相互作用带来的药物不良反应和不良事件,提高患者的依从性。此外,本研究还测定了老年患者服用包含曲马多在内多种药物后的尿液,并观察同时服用的其他药物是否干扰曲马多的SERS检测。
-
BWS415-785H便携式拉曼光谱仪(美国B & W Tek公司);KQ-250DB数控超声波清洗器(昆山市超声仪器有限公司);Vortex-Genie2多功能旋涡混合器(美国 Scientific Industries 公司);TG16-WS 离心机(上海卢湘仪离心机有限公司);电子分析天平(北京赛多利斯仪器系统有限公司);TU-1902 紫外可见分光光度计(北京普析通用仪器有限责任公司);Zeiss EVO MA-10 扫描电子显微镜(德国 Carl-Zeiss 公司)。
-
盐酸曲马多(99.0%,中国食品药品检定研究院);柠檬酸三钠(C6H5NaO7·H2O,国药集团化学试剂有限公司);硼酸钠(Na2B4O7)、氢氧化钾(KOH)、硝酸银(AgNO3)、碘化钾(KI)、硫酸镁(MgSO4)、异丙醇(iPrOH)、氯仿(CHCl3)、甲苯(C7H8) 和乙醚 (C4H10O) (分析纯, 购自上海试剂公司)。
本研究使用了空白尿液及医院患者的真实尿液,取样后立即冷冻并储存于−80℃低温冰箱。医疗期间盐酸曲马多收集时间和使用情况的信息,以及从健康志愿者和患者获得的人尿液样本的分析得到了当地伦理委员会的批准。
-
精密称量盐酸曲马多对照品100 mg,配制成浓度为100 μg/ml的储备液。再利用去离子水按照浓度梯度稀释,依次得到浓度为 10、9、8、7、6、5、4、3、2、1、0.95、0.9、0.85、0.8、0.75、0.7、0.65、0.6、0.55、0.5、0.45 μg/ml的曲马多溶液。
-
按照Lee等[13]的方法制备银胶。在300 ml去离子水中加入54 mg AgNO3并不断加热至微沸,然后加入6 ml质量分数为1%的C6H5NaO7· H2O溶液,继续加热并充分搅拌约1 h,直到溶液变为灰绿色,停止加热,自然冷却至室温,置于棕色玻璃瓶中,避光保存。
-
银胶中加入5 μl KI溶液(1 mol/L),孵育20 min去除银胶表面杂质信号,然后取10 μl胶体与5 μl样品混合,并加入2 μl MgSO4(0.01 mol/L,加入Mg2+目的是团聚银胶颗粒以增强检测信号),最后将混合物加入石英玻璃管中,立即置于拉曼光谱仪中采集光谱。真实尿液实验步骤同空白尿液。
将盐酸曲马多溶解在水中,浓度范围为0.5~100 μg/ml。此外,对掺入空白尿液中的盐酸曲马多使用相同的浓度范围。
-
光谱检测参数如下:激光波长785 nm,分辨率5 cm−1,积分时间3 s,扫描次数1次,激光功率 7 mW。
-
将1 ml尿样与0.05 ml一定浓度的药物溶液混合在4 ml EP管中。然后,用Na2B4O7/KOH缓冲溶液调节pH至7。添加1 ml萃取剂氯仿∶异丙醇(9∶1,V/V)以从尿样中提取曲马多。在室温下剧烈震荡后,以
6000 r/min 离心5 min。弃去上层清液,取下层液0.5 ml至试管里,用氮气仪吹干下层液,加入与下层液等量的去离子水,用于进一步实验。 -
采用BWSpec软件对采集到的光谱原始数据进行初步处理,主要为光谱平滑和基线校正,然后利用 Matlab 软件对数据进行光谱波段的截取,选取300~
1800 cm−1 处的光谱数据进行分析,再采用Origin 8软件对处理好的数据进行绘图。 -
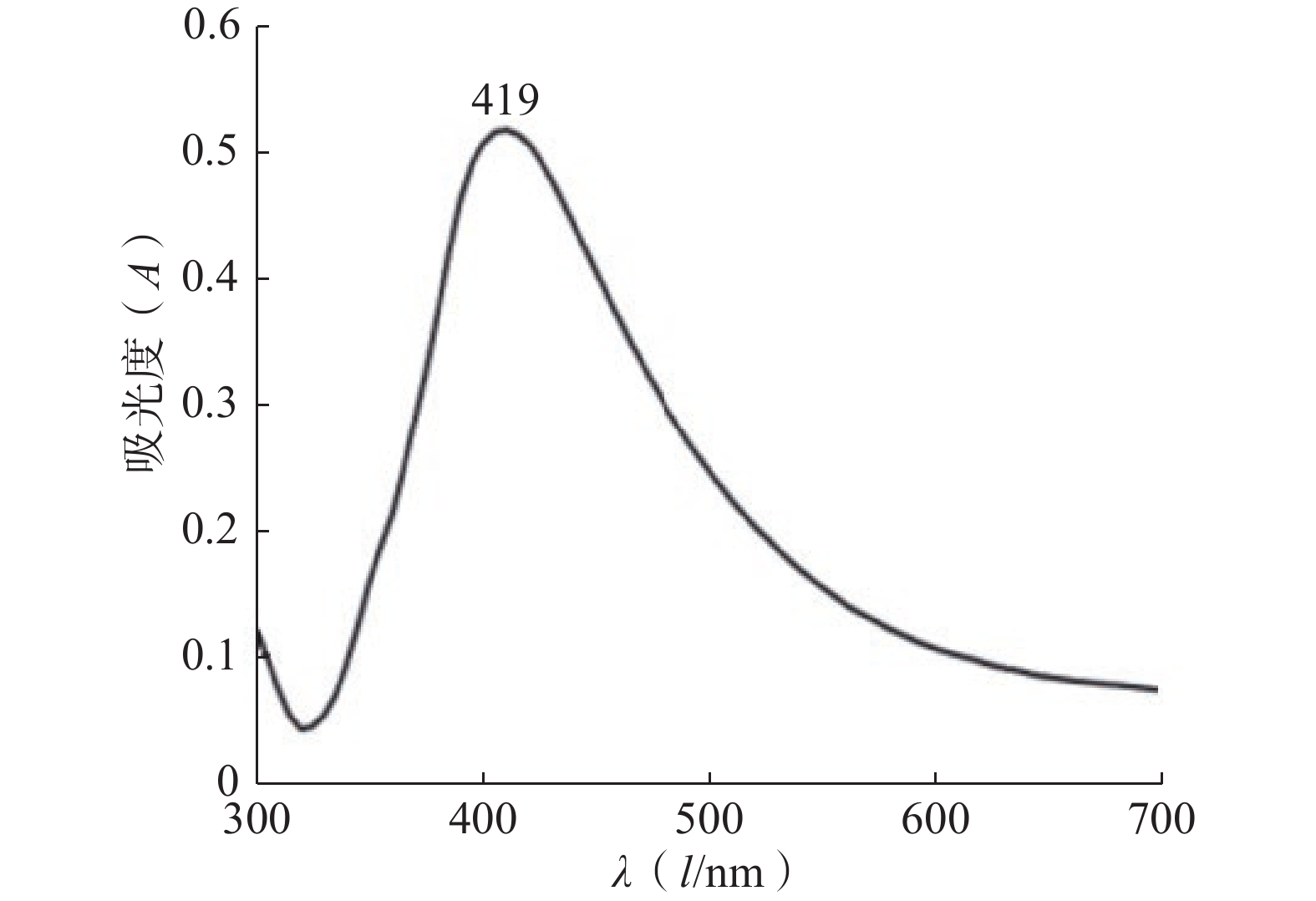
紫外图谱表征:使用紫外分光光度计在 300~700 nm 波长范围内对银胶进行紫外光谱扫描,并观察物质的紫外特征吸收峰。银纳米颗粒的紫外吸收光谱如图1所示。银纳米颗粒的紫外可见吸收光谱仅在419 nm处有一个吸收峰,半峰宽较窄,这反映出银纳米颗粒大小较均匀,具有良好的分散性。
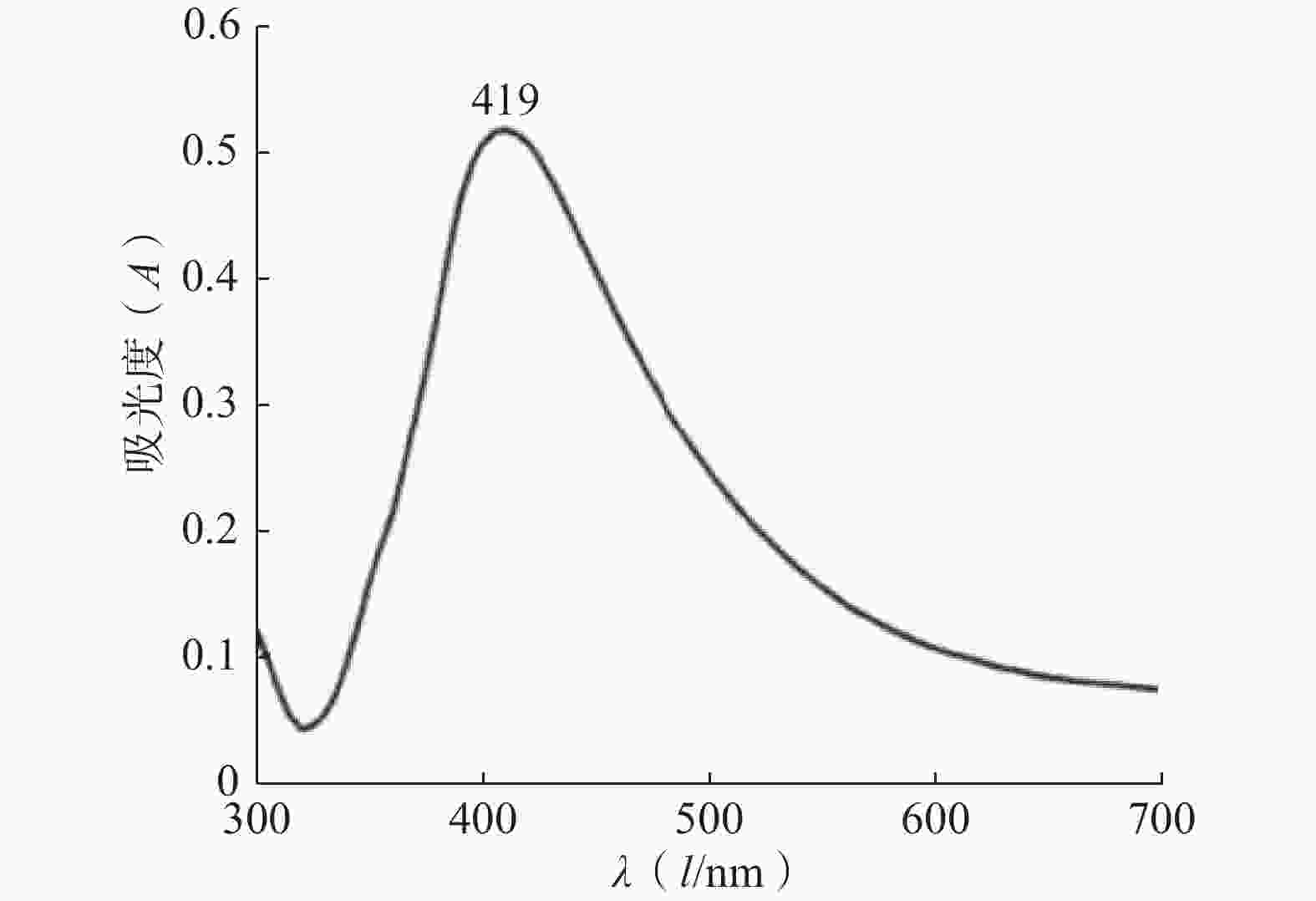
图 1 银胶的紫外可见光吸收光谱图
扫描电镜表征:使用扫描电子显微镜(SEM)扫描纳米银胶颗粒,观察纳米银胶颗粒的形态。银胶颗粒形似球状,大小均匀,直径约为50 nm。
-
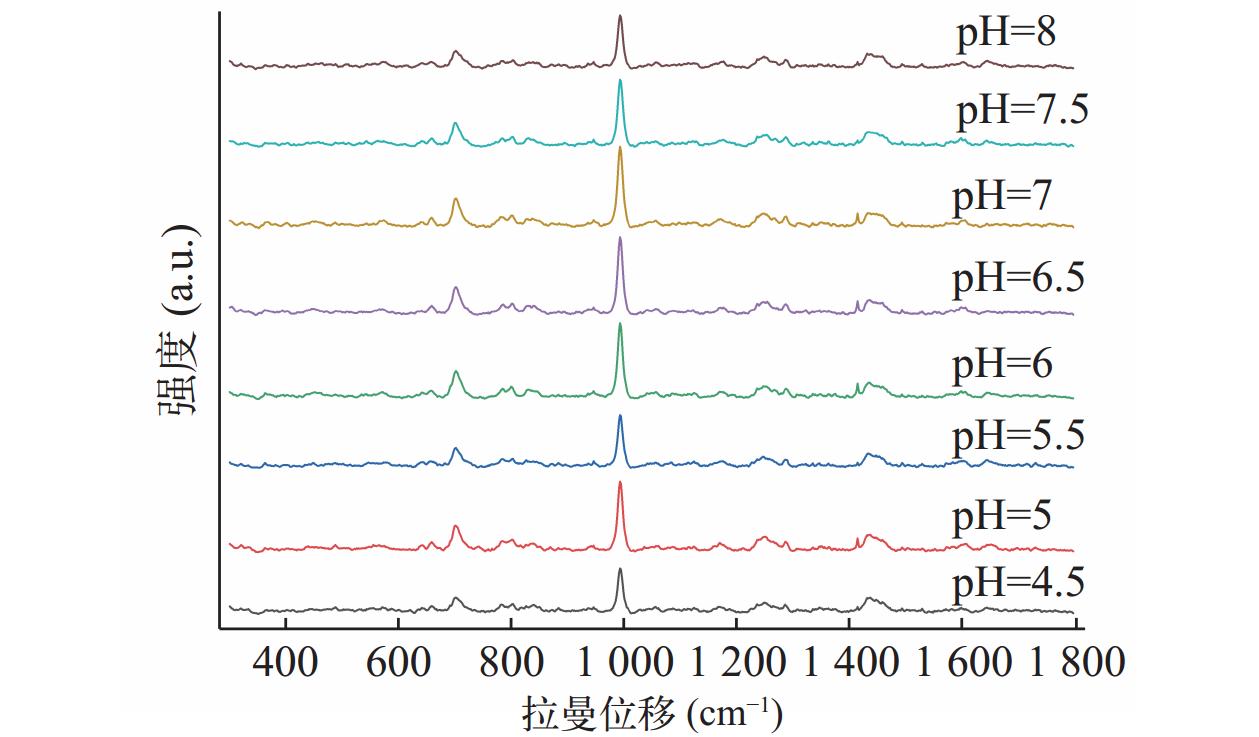
将盐酸曲马多对照品溶于去离子水中,改变溶液的pH值,并评估其对SERS的影响。使用Na2B4O7/KOH缓冲液将溶液pH值调整在4.5~8之间变化,立即获取SERS数据。由图2可知,样本pH 为7.0时,盐酸曲马多SERS信号最强。
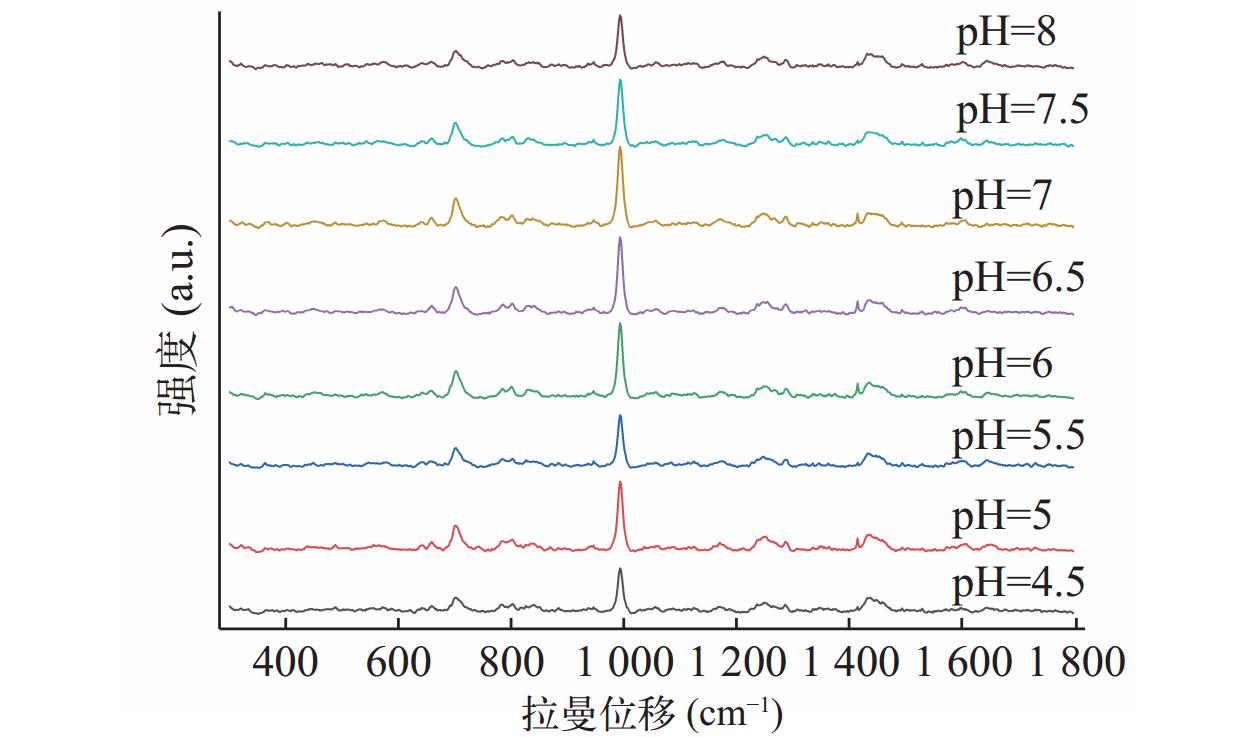
图 2 盐酸曲马多水溶液在pH值为4.5~8的SERS光谱
-
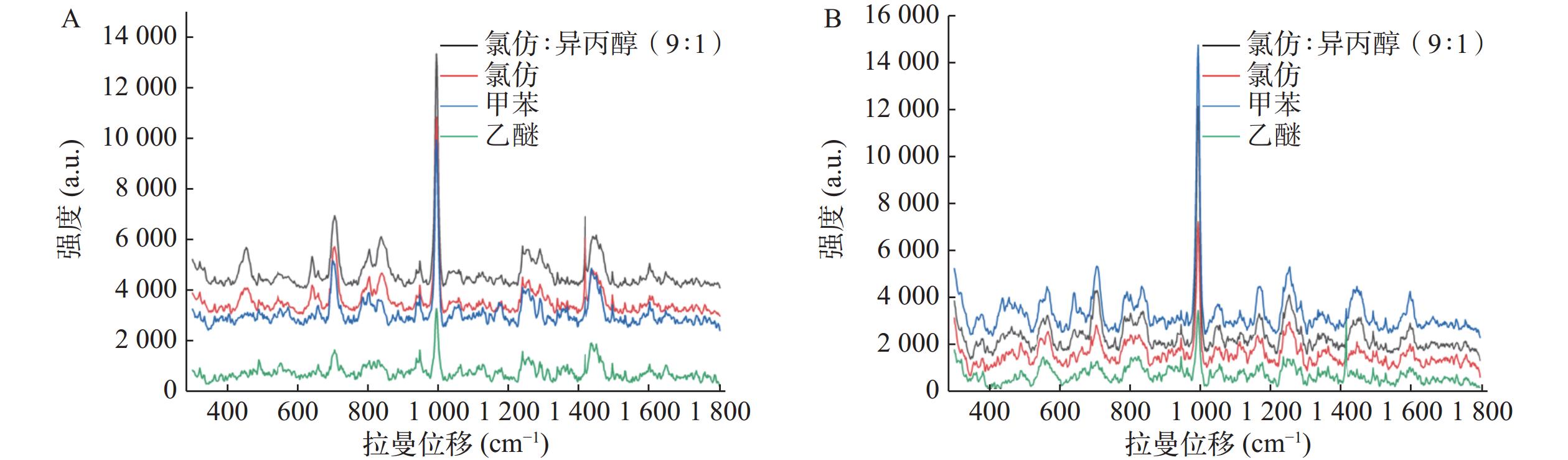
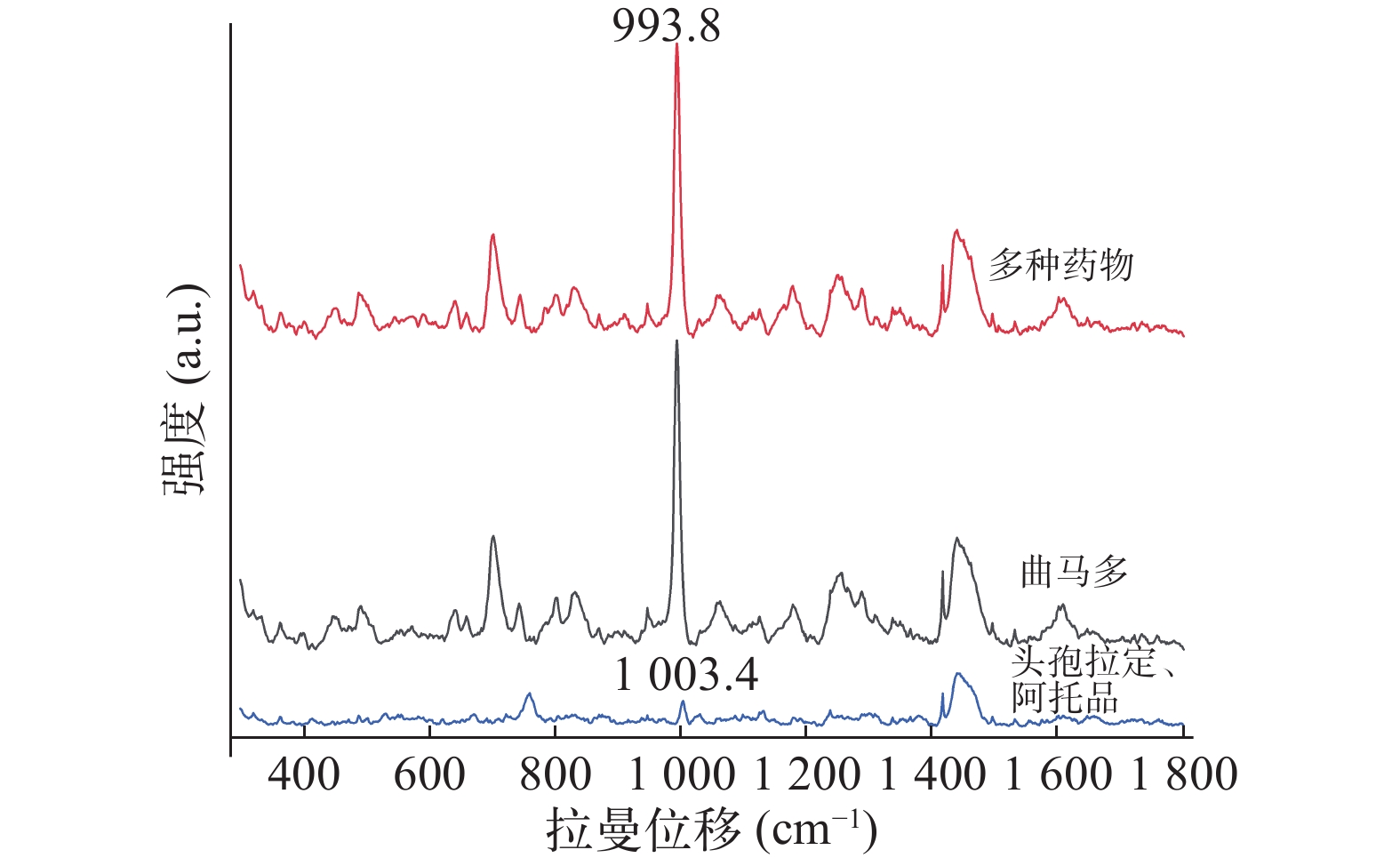
Yu等[6]研究发现氯仿∶异丙醇(9∶1)比氯仿或正丁醇能更有效提取尿液中吗啡。本文评估了氯仿、氯仿∶异丙醇(9∶1)以及甲苯和乙醚提取空白尿液加标和真实尿液的效果,以993.8 cm−1的相对峰强度为指标。SERS检测结果显示空白尿液加标和真实尿液中曲马多993.8 cm−1信号均以氯仿∶异丙醇(9∶1)作为萃取剂时最强,乙醚作为萃取剂时最弱,故选定氯仿∶异丙醇(9∶1)作为曲马多的萃取溶剂(图3)。
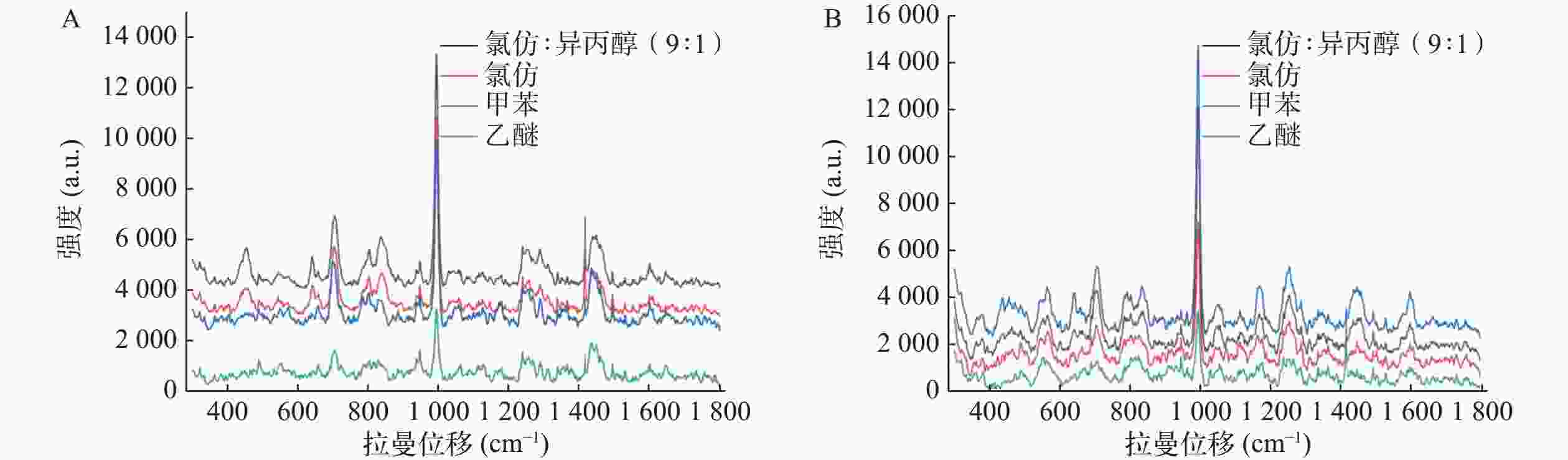
图 3 基于LLE法采用不同溶剂的SERS光谱
-
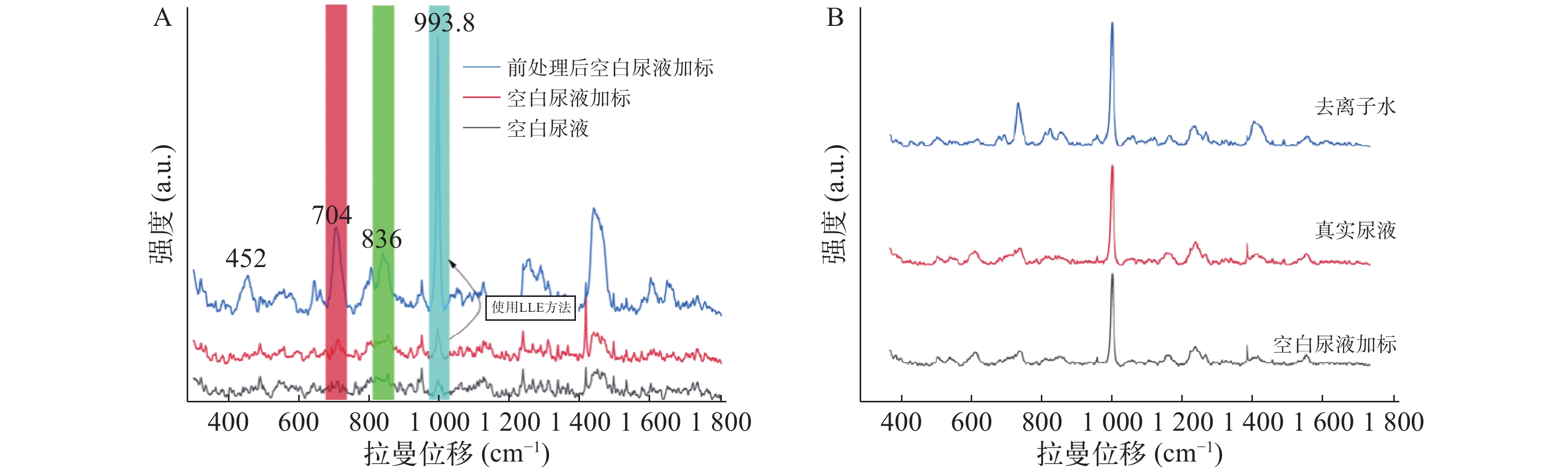
由图4A可见,未经前处理尿样直接进行拉曼光谱仪测定,结果显示空白尿液中尿素分子信号与空白尿液加标的704 cm−1和800~850 cm−1处的拉曼信号部分重叠。该干扰峰是由尿素分子中的肌氨酸酐干扰引起的,因此不应把704 cm−1和800~850 cm−1处作为特征峰。而993.8 cm−1处受尿素分子的拉曼信号干扰较弱,可初步选作快速鉴定尿液中存在曲马多的定性依据。此外,经LLE尿液中曲马多的前处理方法,993.8 cm−1处目标分子的特征峰信号受尿液干扰进一步变弱,信号明确增强。
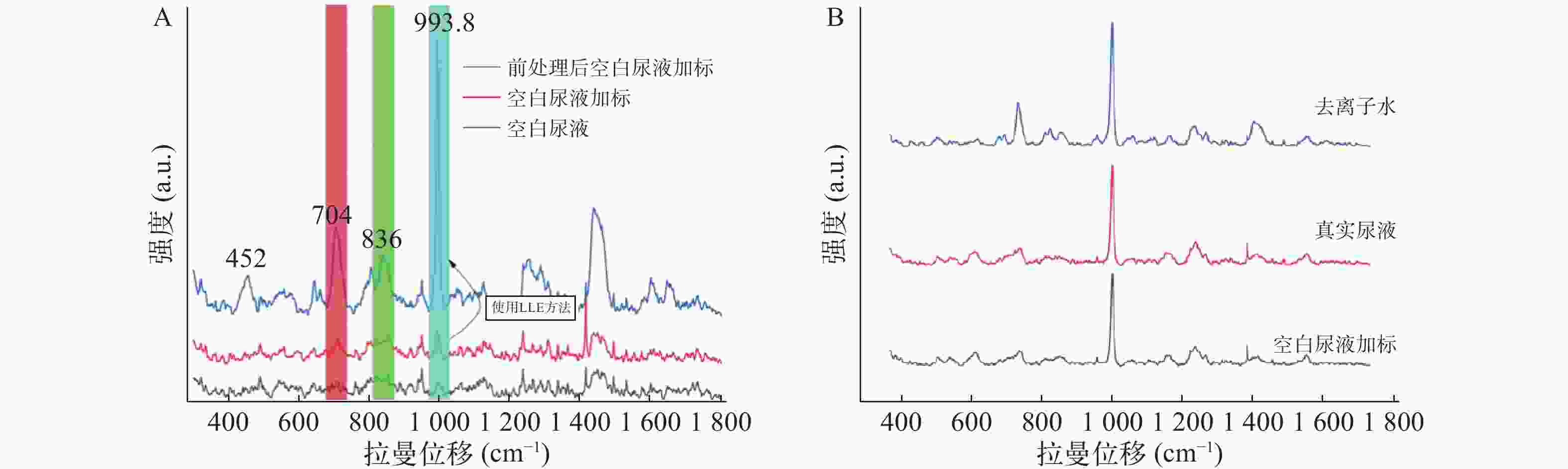
图 4 空白尿液加标与真实尿液的基质成分对曲马多测定的干扰
图4B可见,由于真实尿液与空白尿液的澄清度不如去离子水,影响了检测器对拉曼散射光的感应,其特征峰较水溶液中信号相对较弱。同时空白尿液与真实尿液中其他杂质与曲马多形成竞争关系,共同吸附在银胶金属粒子表面,产生一定抑制作用,对特征峰峰形也有一定的影响。因此经LLE尿液中曲马多的前处理方法,真实尿液中993.8 cm−1特征峰信号最强。
-
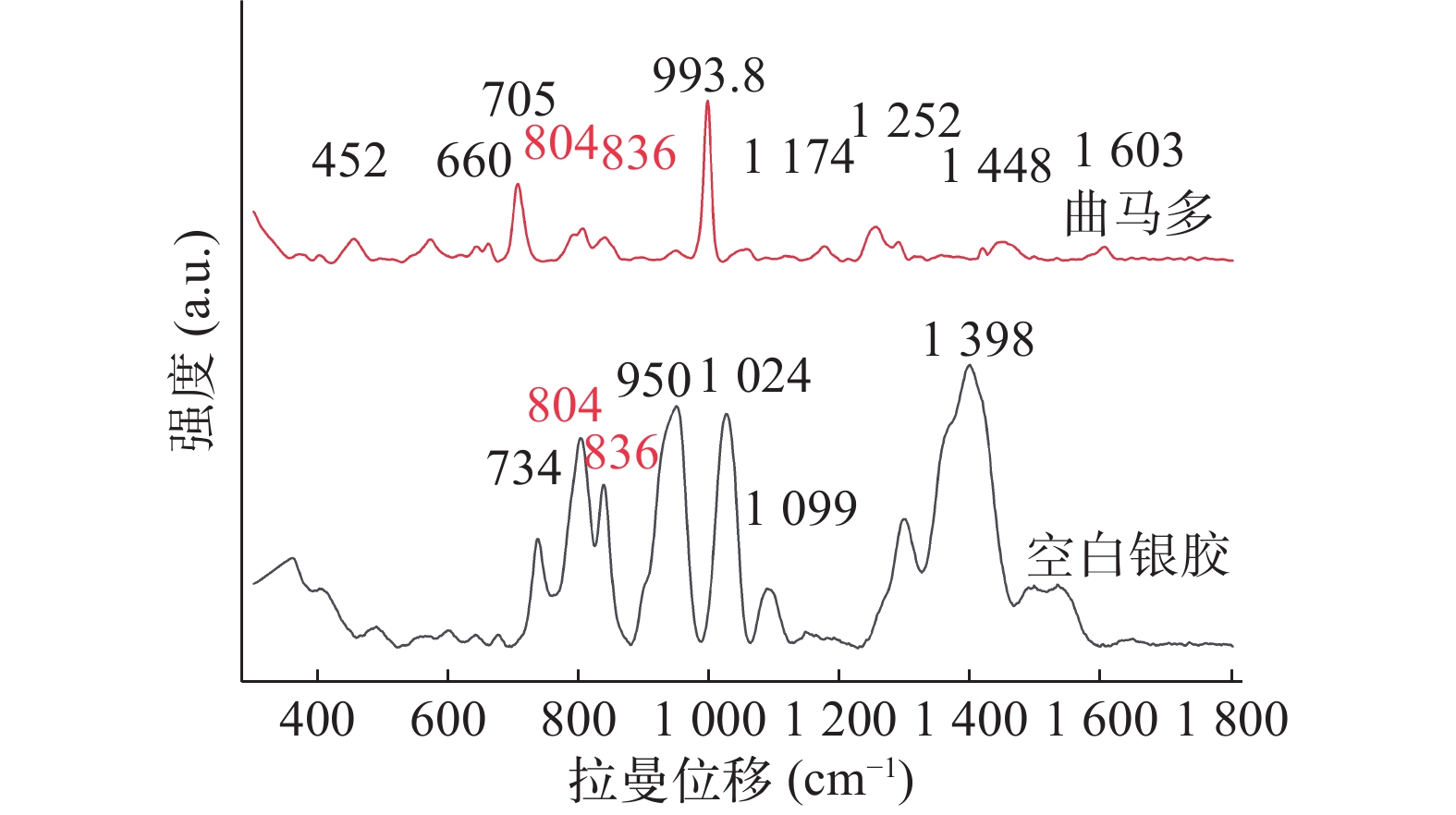
Kimani等[14]研究显示曲马多的 SERS 以 452、660、704、804、836、993.8、
1172、 1253、 1288、 1448 和1603 cm−1 处的峰为主。由图5可得,研究发现804、836与993.8 cm−1在低浓度情况下SERS信号较强,但银胶基底SERS的峰与曲马多804、836 cm−1处峰重合,故本研究以993.8 cm−1 处尿液中曲马多峰为特征峰。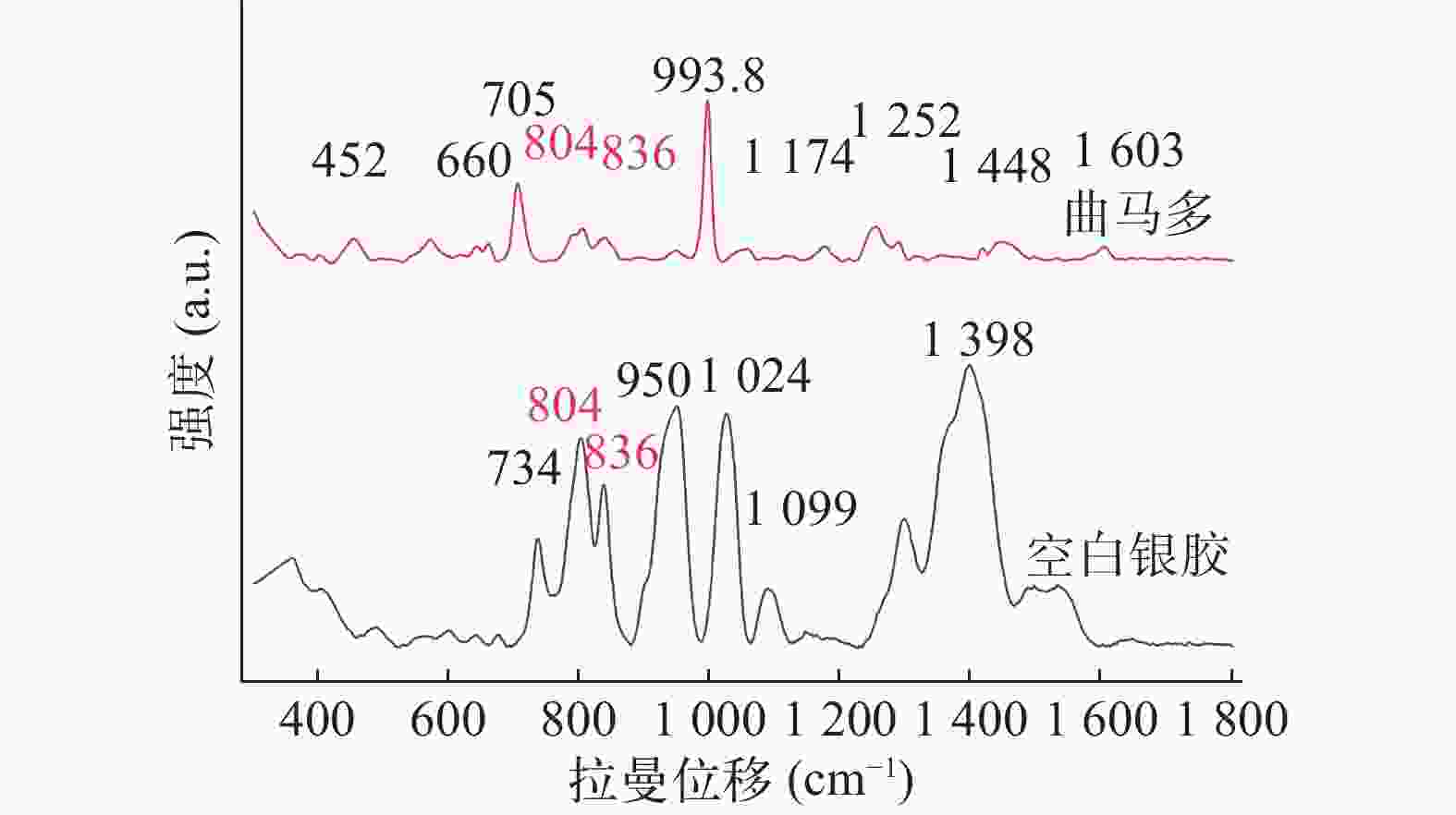
图 5 空白尿液加标与银胶的SERS光谱图
-
经查阅文献,头孢拉定与阿托品均以原形药物形式从尿液排出,且他们的特征峰也在993.8cm−1附近[15-16],这可能会影响曲马多的检测。为了进一步验证以上药物同时服用时对尿液中曲马多的检测是否有干扰,本研究采用LLE 预处理并使用SERS检测空白尿液加头孢拉定、阿托品及曲马多的样本。由图6可知,空白尿液加头孢拉定、阿托品未检测到993.8 cm−1处特征峰,而检测空白尿液加头孢拉定、阿托品及曲马多仍可以清晰地识别出曲马多993.8 cm−1处的特征峰。因此,SERS结合LLE可以实现对同时服用头孢拉定与阿托品的老年患者尿液中曲马多的测定。
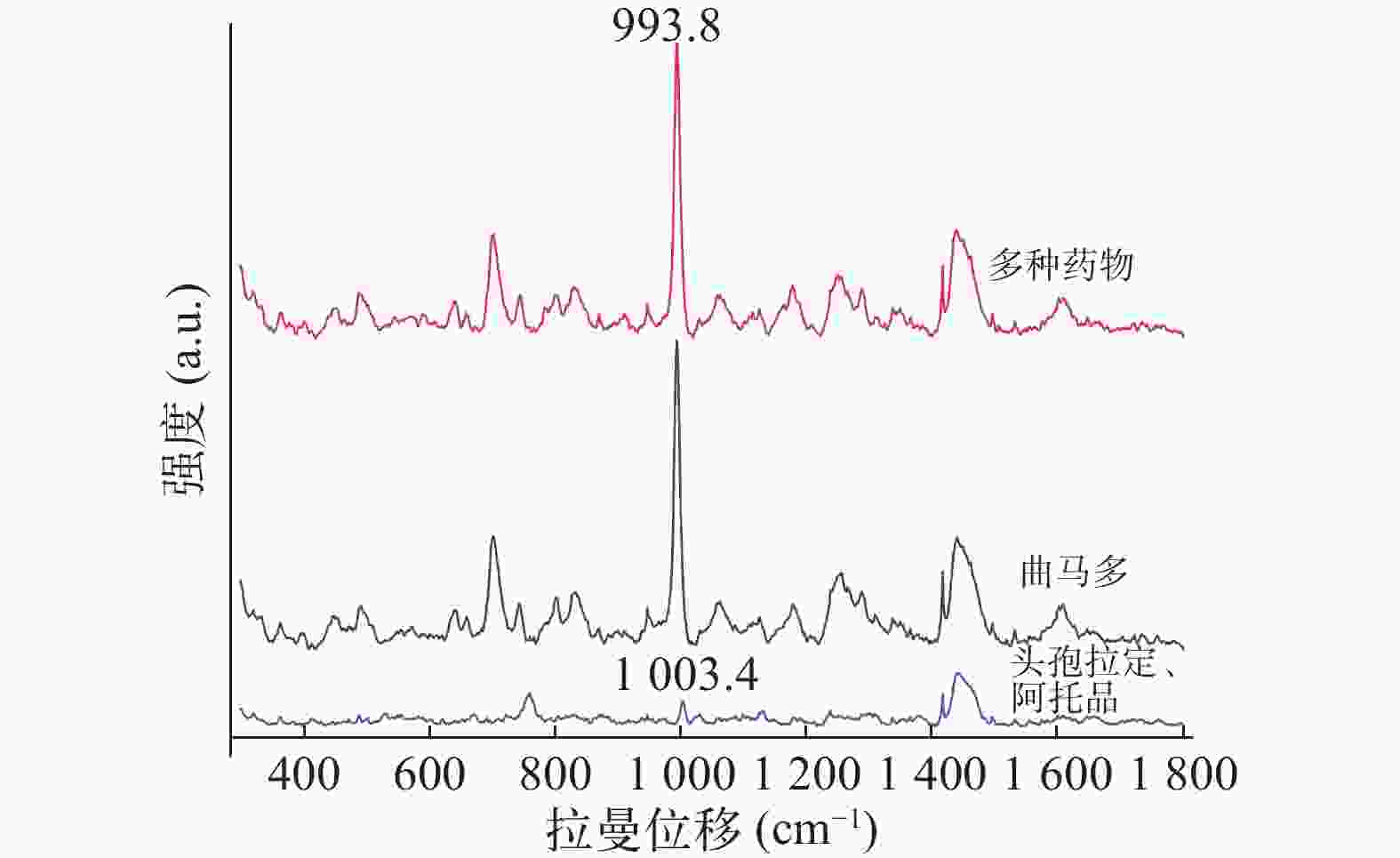
图 6 空白尿液中含曲马多等多种药物及空白尿液中头孢拉定、阿托品的SERS光谱图
-
配制不同浓度空白尿液中盐酸曲马多溶液(1、5、10、20、30、40、50、60、70、80、90、100 μg/ml),将这些溶液置于拉曼光谱仪下的石英玻璃管中进行拉曼光谱测定,以波峰993.8 cm−1的相对强度(Y)为纵坐标,盐酸曲马多对照品溶液浓度(X)为横坐标,拟合线性回归方程为Y=204.35 X−465.62,回归系数r=
0.9952 ,盐酸曲马多的浓度在1~100 μg/ml范围内与特征峰993.8 cm−1的相对强度呈良好线性关系。取上述盐酸曲马多对照品溶液逐级稀释测定,测得盐酸曲马多的定量限(S/N=10)和检测限(S/N=3)分别为 1.54 μg/ml和0.53 μg/ml。
-
取“3.4.2”项下曲马多分别为 5、50、80 μg/ml的加标溶液,分别于1 d和1周内重复测定6次,测得日内、日间的精密度(RSD)及准确度(RE)见表1。结果表明,低、中、高含量的日内、日间的精密度及准确度均良好。
表 1 曲马多不同浓度的回收率、精密度及准确度
浓度
(μg/ml)平均
回收率
(n=3)RSD
(n=5)日内精密度(n=6) 日间精密度(n=6) RSD(%) RE(%) RSD(%) RE(%) 5 93.01 3.92 3.38 −1.47 4.61 −4.03 50 96.5 4.27 3.33 −2.64 4.68 4.67 80 94.9 4.68 3.54 −2.92 4.65 −4.91 -
取“3.4.2”项下标准品液配制低、中、高( 5、50、80 μg/ml)3个浓度的加标溶液,每个浓度3份供试样品,按“2.3”项下处理并用SERS测定,测得结果与实际浓度比值计算回收率,见表1。
-
同回收率试验,配制3个浓度的盐酸曲马多加标溶液,于−25℃冷冻后,按“2.3”项下处理并用SERS测定,每个样本重复冻融5次,计算RSD,见表1。
-
通过检测3位服用曲马多患者的尿液样本,SERS分析结果可以清楚地识别出患者尿液中曲马多的特征峰。根据标准曲线可得患者A、B、C尿液中曲马多浓度分别为39.55、33.74、27.57 μg/ml。因此,SERS结合LLE可以实现对服用曲马多者尿液中曲马多的快速、灵敏的测定。
-
对比Alharbi等[10]的研究使用人工尿液做模拟试验,本研究进一步对临床上服用曲马多患者的真实尿液进行检测,采用LLE前处理方法降低尿液中成分对背景 SERS 信号的影响,曲马多的检测限与之前的研究相当(530 ng/ml),证明了本方法检测真实尿液中曲马多的有效性,且方法简单、高效、经济,可以用于曲马多个性化用药的定性和定量分析,并进一步扩展至其他药物的监测。
Rapid determination of tramadol in urine by surface-enhanced Raman spectroscopy
-
摘要:
目的 应用液液萃取(LLE)-表面增强拉曼光谱(SERS),建立快速检测尿液中曲马多的方法。 方法 以氯仿∶异丙醇(9∶1)萃取剂从尿液中提取曲马多,采用增强拉曼光谱(波长为785 nm)检测尿液样品中的曲马多。 结果 曲马多的定量曲线Y=204.35 X−465.62,r= 0.9952 ,线性范围为1~100 μg/ml,该方法曲马多的检测限(S/N=3)为0.53 μg/ml;与常规方法相比,SERS具有高灵敏度及合理的重现性。结论 此方法简单、高效、经济,可以用于曲马多个性化用药的定性和定量分析。 Abstract:Objective To establish a method for rapid detection of tramadol in urine by liquid-liquid extraction(LLE)-surface-enhanced Raman spectroscopy (SERS). Methods Tramadol was extracted from urine with chloroform∶isopropyl alcohol (9∶1) extractant and detected in urine samples by enhanced Raman spectroscopy (wavelength 785 nm). Results The quantitative curve of tramadol was Y=204.35 X−465.62, r= 0.9952 , and the linear range was 1-100 μg/ml. The detection limit of tramadol by this method (S/N=3) was 0.53 μg/ml. The sensitivity of SERS was higher than that of conventional methods, and it had reasonable reproducibility.Conclusion This method is simple, efficient and economical, and can be used for qualitative and quantitative analysis of tramadol personalized medicine. -
表 1 曲马多不同浓度的回收率、精密度及准确度
浓度
(μg/ml)平均
回收率
(n=3)RSD
(n=5)日内精密度(n=6) 日间精密度(n=6) RSD(%) RE(%) RSD(%) RE(%) 5 93.01 3.92 3.38 −1.47 4.61 −4.03 50 96.5 4.27 3.33 −2.64 4.68 4.67 80 94.9 4.68 3.54 −2.92 4.65 −4.91  下载: 导出CSV
下载: 导出CSV
-
[1] MOULIS F, ROUSSEAU V, ABADIE D, et al. Serious adverse drug reactions with tramadol reported to the French pharmacovigilance database between 2011 and 2015[J]. Therapie, 2017, 72(6):615-624. doi: 10.1016/j.therap.2017.03.004 [2] TIAN F Y, YANG R N, CHEN Z Y, et al. The prevalence and factors associated with potentially inappropriate medication use in Chinese older outpatients with cancer with multimorbidity[J]. J Geriatr Oncol, 2022, 13(5):629-634. doi: 10.1016/j.jgo.2022.02.006 [3] EL-SAYED A A Y, MOHAMED K M, NASSER A Y, et al. Simultaneous determination of tramadol, O-desmethyltramadol and N-desmethyltramadol in human urine by gas chromatography–mass spectrometry[J]. J Chromatogr B, 2013, 926:9-15. doi: 10.1016/j.jchromb.2013.02.019 [4] CHENG P S, LEE C H, LIU C, et al. Simultaneous determination of ketamine, tramadol, methadone, and their metabolites in urine by gas chromatography-mass spectrometry[J]. J Anal Toxicol, 2008, 32(3):253-259. doi: 10.1093/jat/32.3.253 [5] HEJABRI KANDEH S, AMINI S, EBRAHIMZADEH H. Simultaneous trace-level monitoring of seven opioid analgesic drugs in biological samples by pipette-tip micro solid phase extraction based on PVA-PAA/CNT-CNC composite nanofibers followed by HPLC-UV analysis[J]. Mikrochim Acta, 2021, 188(8):275. doi: 10.1007/s00604-021-04931-w [6] YU B R, CAO C T, LI P, et al. Sensitive and simple determination of zwitterionic morphine in human urine based on liquid-liquid micro-extraction coupled with surface-enhanced Raman spectroscopy[J]. Talanta, 2018, 186:427-432. doi: 10.1016/j.talanta.2018.04.094 [7] ALMASOUD N, ALOMAR T S, XU Y, et al. Rapid detection and quantification of paracetamol and its major metabolites using surface enhanced Raman scattering[J]. Analyst, 2023, 148(8):1805-1814. doi: 10.1039/D3AN00249G [8] ZHU Q X, LI X H, LI D, et al. A rapid therapeutic drug monitoring strategy of carbamazepine in serum by using coffee-ring effect assisted surface-enhanced Raman spectroscopy[J]. Molecules, 2022, 28(1):128. doi: 10.3390/molecules28010128 [9] ZHU Q X, YU X Y, WU Z B, et al. Antipsychotic drug poisoning monitoring of clozapine in urine by using coffee ring effect based surface-enhanced Raman spectroscopy[J]. Anal Chim Acta, 2018, 1014:64-70. doi: 10.1016/j.aca.2018.02.027 [10] ALHARBI O, XU Y, GOODACRE R. Detection and quantification of the opioid tramadol in urine using surface enhanced Raman scattering[J]. Analyst, 2015, 140(17):5965-5970. doi: 10.1039/C5AN01177A [11] MARKINA N E, MARKIN A V, WEBER K, et al. Liquid-liquid extraction-assisted SERS-based determination of sulfame-thoxazole in spiked human urine[J]. Anal Chim Acta, 2020, 1109:61-68. doi: 10.1016/j.aca.2020.02.067 [12] DOCTOR E L, MCCORD B. Comparison of aggregating agents for the surface-enhanced Raman analysis of benzodiazepines[J]. Analyst, 2013, 138(20):5926-5932. doi: 10.1039/c3an00669g [13] LEE P C, MEISEL D. Adsorption and surface-enhanced Raman of dyes on silver and gold sols[J]. J Phys Chem, 1982, 86(17):3391-3395. doi: 10.1021/j100214a025 [14] KIMANI M M, LANZAROTTA A, BATSON J S. Trace level detection of select opioids(fentanyl, hydrocodone, oxycodone, and tramadol)in suspect pharmaceutical tablets using surface-enhanced Raman scattering(SERS)with handheld devices[J]. J Forensic Sci, 2021, 66(2):491-504. doi: 10.1111/1556-4029.14600 [15] 韩斯琴高娃, 沙轩宇, 赵航, 等. 利用表面增强拉曼光谱技术检测硫酸阿托品[J]. 中国药师, 2017, 20(12):2277-2281. [16] 范蕾, 张雁. 头孢拉定中三种有关物质的表面增强拉曼光谱研究[J]. 中国药师, 2014, 17(7):1089-1093. -

 点击查看大图
点击查看大图
图(6) / 表(1)
计量
- 文章访问数: 21898
- HTML全文浏览量: 5688
- PDF下载量: 34
- 被引次数: 0
