

 下载:
下载:

-
叶绿体是植物体中具有独立遗传特性的一种细胞器,由于它具有基因组结构简单和母系遗传等特点,引起了广泛关注[1-2]。典型植物叶绿体基因组为双链环状结构,大小通常介于100~250 kb之间[3]。在陆地植物中,该基因组呈现高度保守性特征,其结构通常包含两个约25 kb的反向重复序列(IR区),以及被IR区分隔的小单拷贝区(SSC,18~20 kb)和大单拷贝区(LSC,81~90 kb)。基因组内一般编码120~130个基因,主要涉及光合作用、遗传信息转录与翻译等核心生物学功能[4]。尽管叶绿体基因组信息量远低于核基因组,但其稳定性和保守性使其成为解析植物系统进化关系、物种分类鉴定及遗传转化研究的重要分子标记,尤其在药用植物资源开发中已展现出广泛应用价值[5]。
丹参(Salvia miltiorrhiza Bunge)是中医临床治疗心脑血管疾病的常用药物之一,具有活血调经、凉血消痈和清心安神的作用,既是一种常用大宗中药材,也是一味妇科良药,有“一味丹参、功同四物”的美誉。丹参为唇形科鼠尾草属植物,鼠尾草属约有1 000种植物,主要分布于中美洲、南美洲、亚洲西南部、地中海以及东亚地区。东亚地区约有100种鼠尾草属植物,其中83种为中国特有。通过检索叶绿体基因组综合数据库(CGIR,https://ngdc.cncb.ac.cn/cgir/)和美国国家生物技术信息中心(NCBI,https://www.ncbi.nlm.nih.gov/),截止2023年10月,共有76种鼠尾草属植物公布了125个叶绿体基因组,包括丹参及其他21种药用植物。通过对已公开发表的所有鼠尾草属植物的叶绿体基因组进行整理和系统分析,发现已公布的鼠尾草属植物叶绿体基因组存在序列碱基缺失、模糊、起始位点不准确等问题。这些问题可能对该属植物的后续研究带来影响。
本研究对125个鼠尾草属植物的叶绿体基因组重新进行了注释分析以确保注释结果的统一性和准确性。通过对鼠尾草属植物叶绿体基因组的密码子偏好性进行分析,为叶绿体基因结构分析等提供支持。
-
检索CGIR和NCBI,对已公布的鼠尾草属叶绿体基因组信息进行收集整理,通过TBtools软件批量下载鼠尾草属叶绿体基因组序列。
-
通过使用基因局部比对工具BLAST v2.12.0[6]进行叶绿体基因组的反向重复区域(IR区)寻找,同时进行大单拷贝区(LSC)和小单拷贝区(SSC)的长度计算。
-
在Linux操作系统中安装叶绿体注释工具CPGAVAS2命令行版[7],对收集整理的125个鼠尾草属叶绿体基因组进行重新注释,并用注释校正工具Apollo v1.11.8软件[8]对注释结果进行手动校正处理。
-
对注释得到的125个鼠尾草属叶绿体基因组的编码序列(CDS)进行提取并去除长度<300 bp(长度<300 bp的基因不适合进行有效密码子计算)和重复的CDS序列,然后使用CodonW 1.4.2对叶绿体编码基因的同义密码子相对使用度(RSCU)进行计算和分析,选择默认参数。
-
在GitHub(https://github.com/)中找到并下载Misa.pl脚本和配置文件Misa.isi,并对Misa.isi中的参数进行设置:单核苷酸重复≥8次,双核苷酸重复≥4次,3核苷酸重复≥4次,4、5、6核苷酸重复≥3次,两个简单重复序列(SSR)之间的序列长度设置为0。在Liunx操作系统中上传Misa.pl脚本,并在同一目录下添加Misa.isi,然后用$ misa.pl filename命令对125个鼠尾草属叶绿体基因组进行SSR分析。
-
从GitHub中下载叶绿体边界区域分析软件IRscope的源代码IRscope V 0.1.R,用Rstudio打开并安装相应的安装包,随后运行IRscope V0.1.R完成IRscope的本地化。分批上传鼠尾草属植物叶绿体基因组注释分析中所得的gb文件,完成对125个鼠尾草属植物叶绿体基因组IR边界的收缩与扩张分析。
-
编写python脚本,在Liunx服务器上将鼠尾草属植物叶绿体基因组注释所得gb文件转化为GBF_MVISTA_ANNOTATION文件用于mVISA数据提交。利用在线软件mVISTA(https://genome.lbl.gov/vista/mvista/submit.shtml)[9]以药用植物丹参(Salvia miltiorrhiza Bge.)叶绿体基因组为参照对125个鼠尾草属植物进行全基因组分析。
-
将鼠尾草属植物叶绿体基因组注释得到的gb文件经修改上传到PhyloSuite v1.2.2软件中,提取125个鼠尾草属植物叶绿体基因组和羽叶枝子花(Dracocephalum palmatum Stephan ex Willd.)以及罗勒(Ocimum basilicum L.)两个外类群的全部CDS序列,手动删除多拷贝和非共有基因。对筛选所得的共有蛋白质编码序列,进行多重序列比对和优化。然后使用IQtree v2.0.6[10]基于最适核苷酸替换模型进行自引导1 000次重复的最大似然法(ML)分析,并利用在线网站iTOL对ML树进行可视化。
-
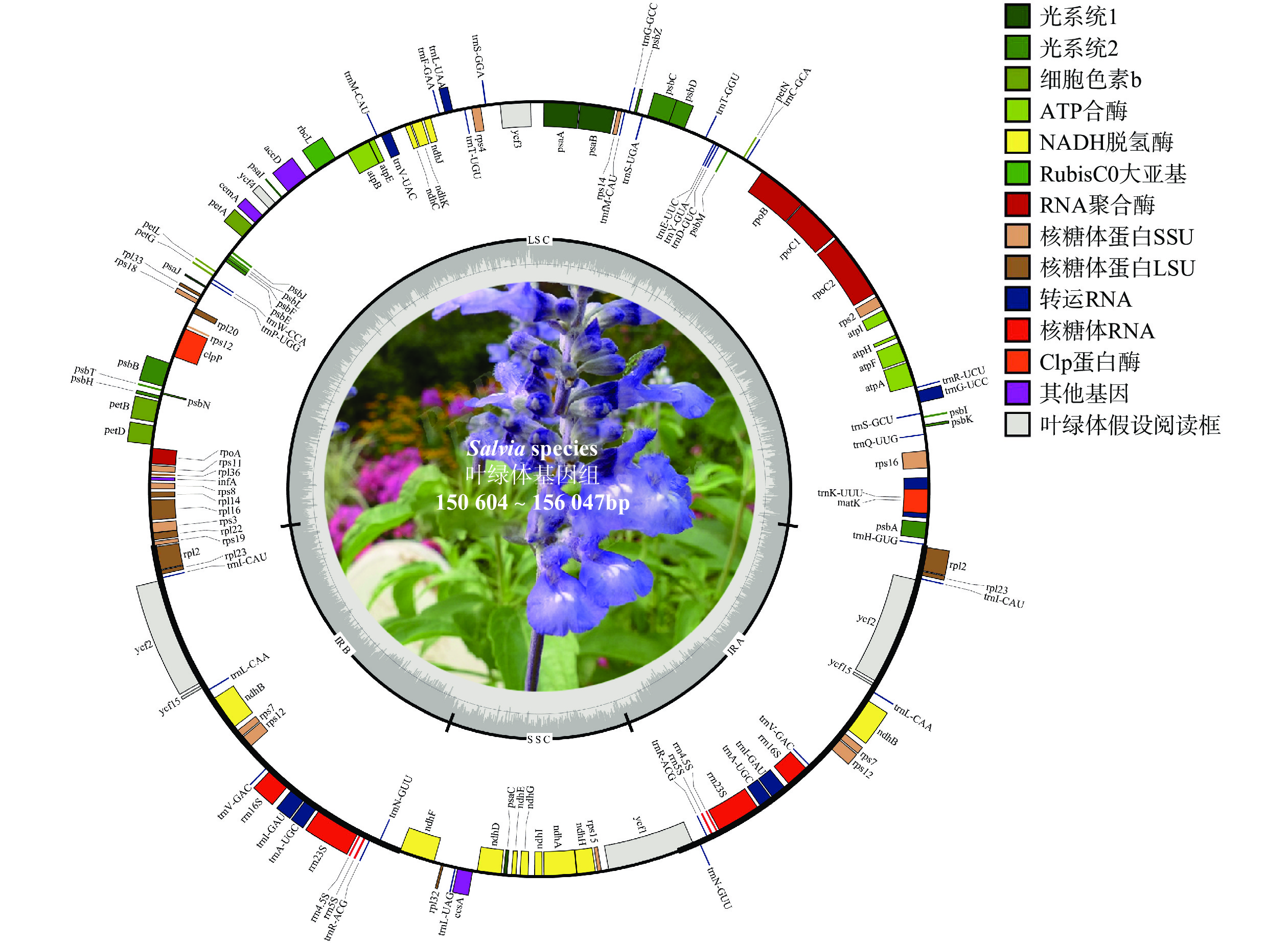
鼠尾草属植物叶绿体基因组表现出典型的环状四分体结构,具有高度相似的基因顺序和基因组结构。基因组大小范围从
150604 bp(Salvia splendens,OM617847)到156047 bp(Salvia splen-dens,OP645384)不等(图1)。LSC区域长度在79 550~84 102 bp之间,SSC区域长度在15 015~19 037 bp之间,且这两个区域被两个IR区域(24 975~30 741 bp)隔开。此外,鼠尾草属植物叶绿体基因组总GC%含量差异较小,GC%含量最小的为37.83%(Salvia splendens,OP645384),最大的为38.13%(Salvia castanea f. castanea,MT634150)。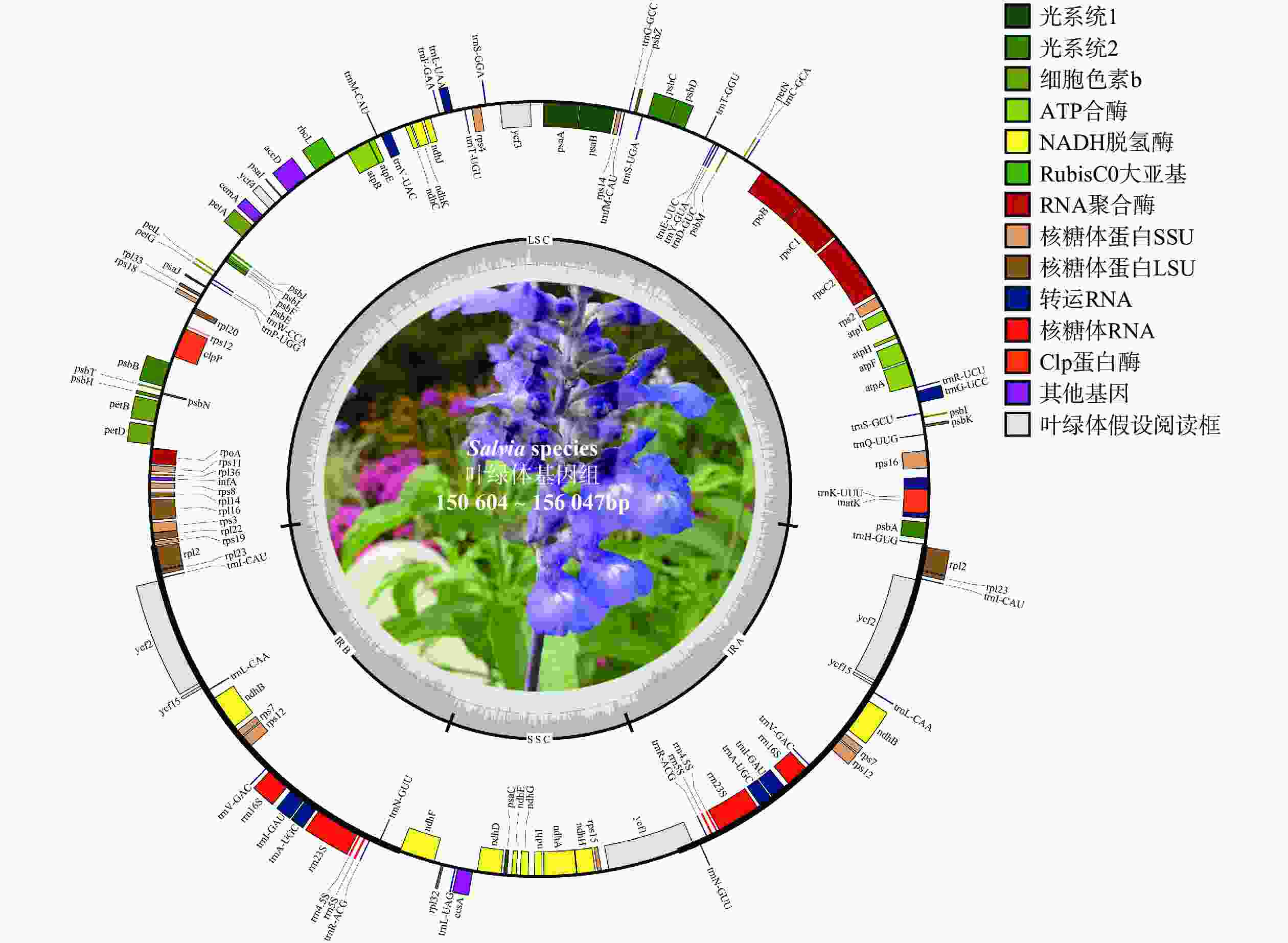
图 1 鼠尾草属植物叶绿体基因组示意图
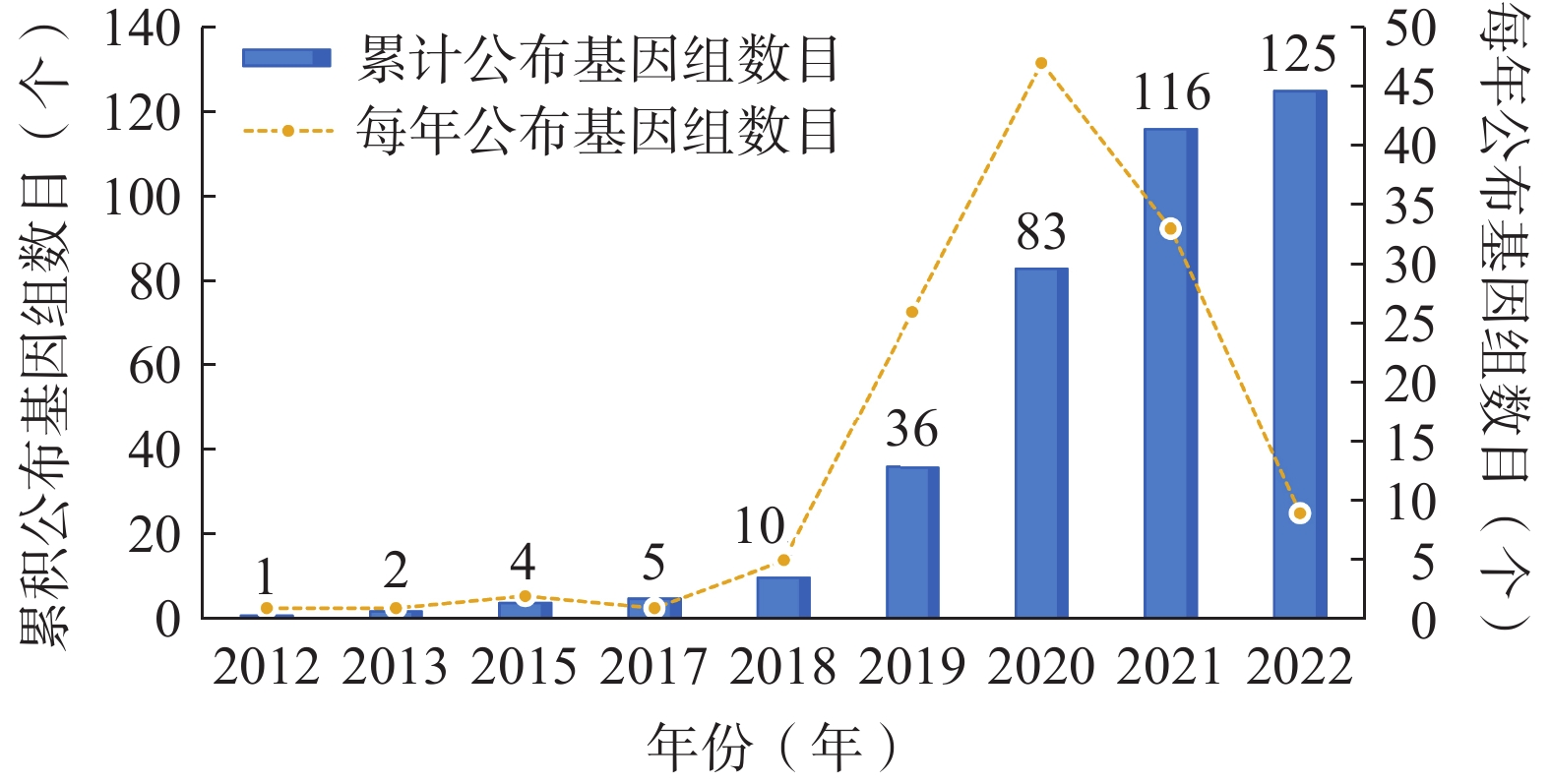
此外,对鼠尾草属植物叶绿体基因组公布年份、每年公布基因组数目、累积公布基因组数目等信息进行统计,结果如图2所示。2012−2018年间,鼠尾草属植物叶绿体基因组信息研究进展缓慢,但自2018年以来,大量叶绿体基因组数据被组装并释放。
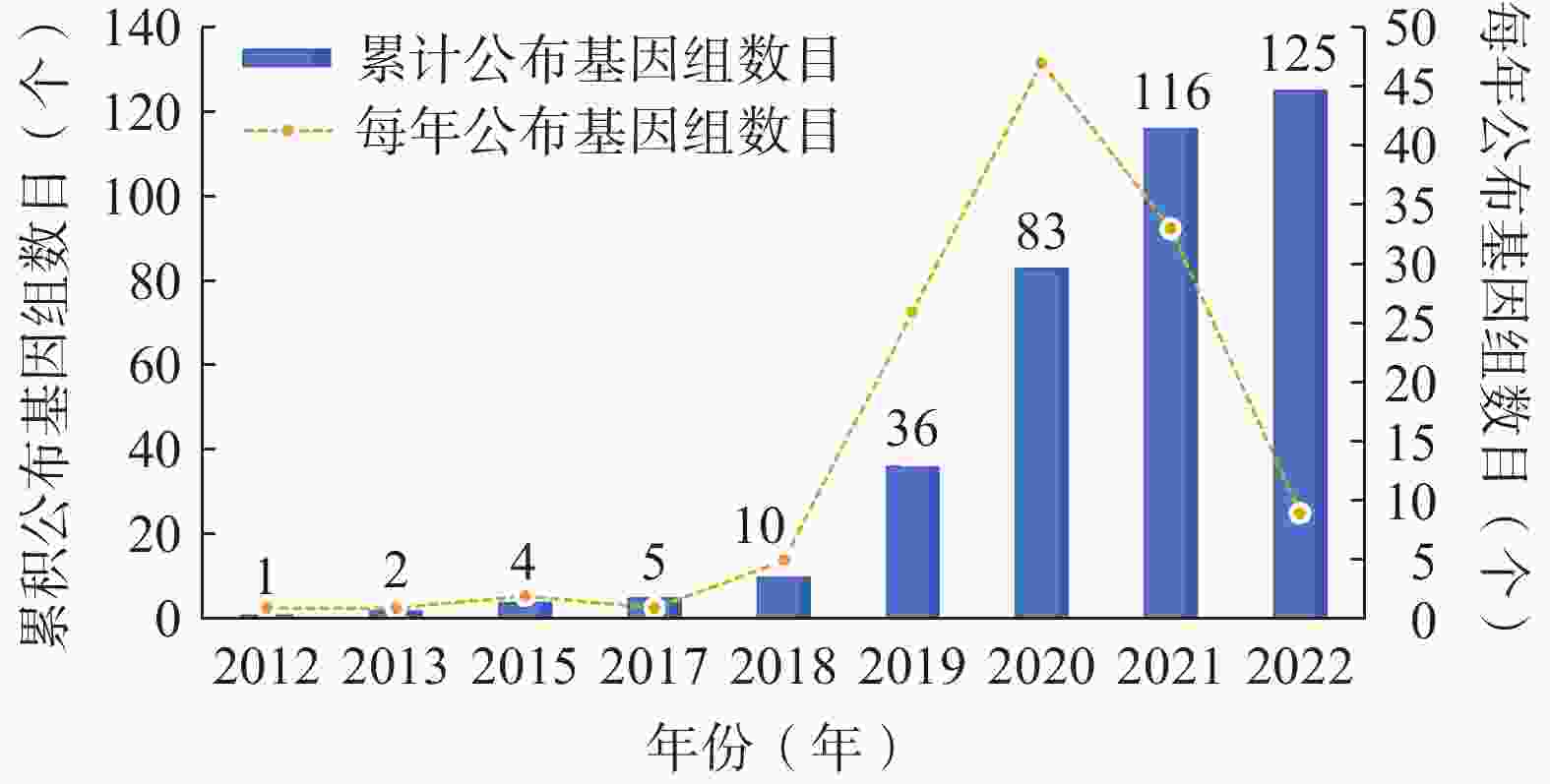
图 2 鼠尾草属植物叶绿体基因组公布年份和累计公布数目统计
-
鼠尾草属植物叶绿体基因按照功能可分为3大类,包括自我复制基因、光合作用基因和其他基因。这些基因中共有17个基因含有内含子,其中9个蛋白质编码基因和6个tRNA基因包含1个内含子,2个基因(ycf3,clpP)包含2个内含子。
此外,在鼠尾草属植物叶绿体基因组的注释分析过程中,发现部分鼠尾草属植物存在tRNA和CDS基因缺失现象。Salvia sonchifolia(MN062355、MW752201和NC_064054)植物的叶绿体基因组缺少trnG-UCC基因、Salvia deserta(MT156378)缺少trnS-UGA基因但多了trnS-GCU基因、Salvia sp. HW-2020(MT634149)缺少trnG-UCC基因、Salvia japonica(NC_035233)缺少trnV-UAC基因、Salvia nanchuanensis(NC_058851)缺少trnS-GCU和trnH-GUG基因、Salvia chienii(OK094518)缺少trnH-GUG基因,而Salvia leucantha(MT156367)和Salvia splendens(OP645384)存在ndhF基因的缺失,Salvia grandifolia(MT156379)存在ycf15基因缺失现象。
-
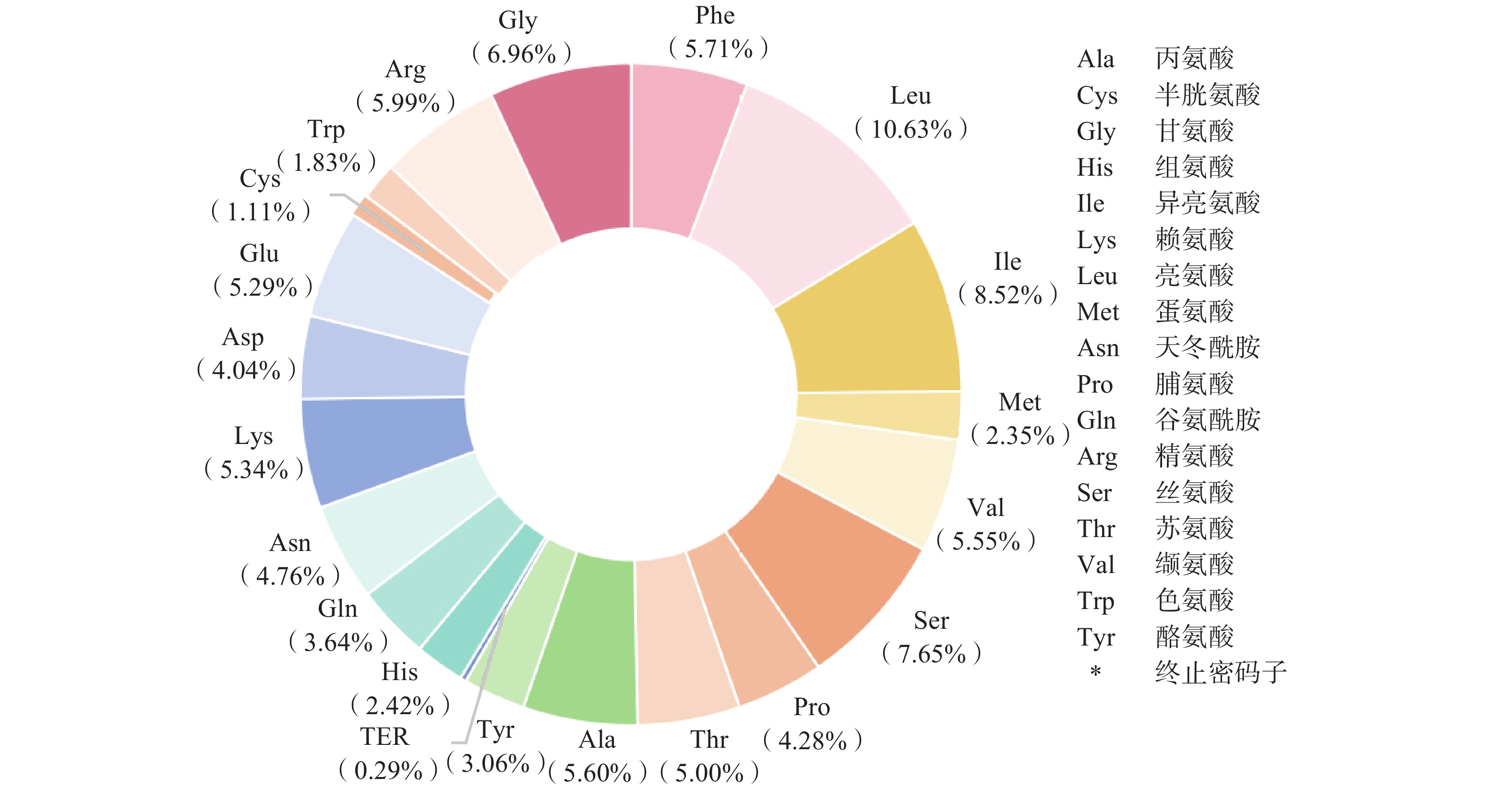
密码子偏好性是生物体进化的重要特征,对基因表达和功能产生影响[11]。鼠尾草属植物叶绿体基因组编码蛋白质的密码子数量在20 334个(Salvia leucantha,MT156367)到23 373个(Salvia filicifolia,MZ677489)之间,共有21种氨基酸和64种密码子(图3、图4)。在这些密码子中,编码亮氨酸(Leu)的密码子数量占比最多,为整个鼠尾草属植物叶绿体基因组的10.63%;其次是异亮氨酸(Ile)和丝氨酸(Ser),分别占比8.52%和7.65%;半胱氨酸是占比最少的氨基酸,所占比例只有1.11%(终止密码子除外)。
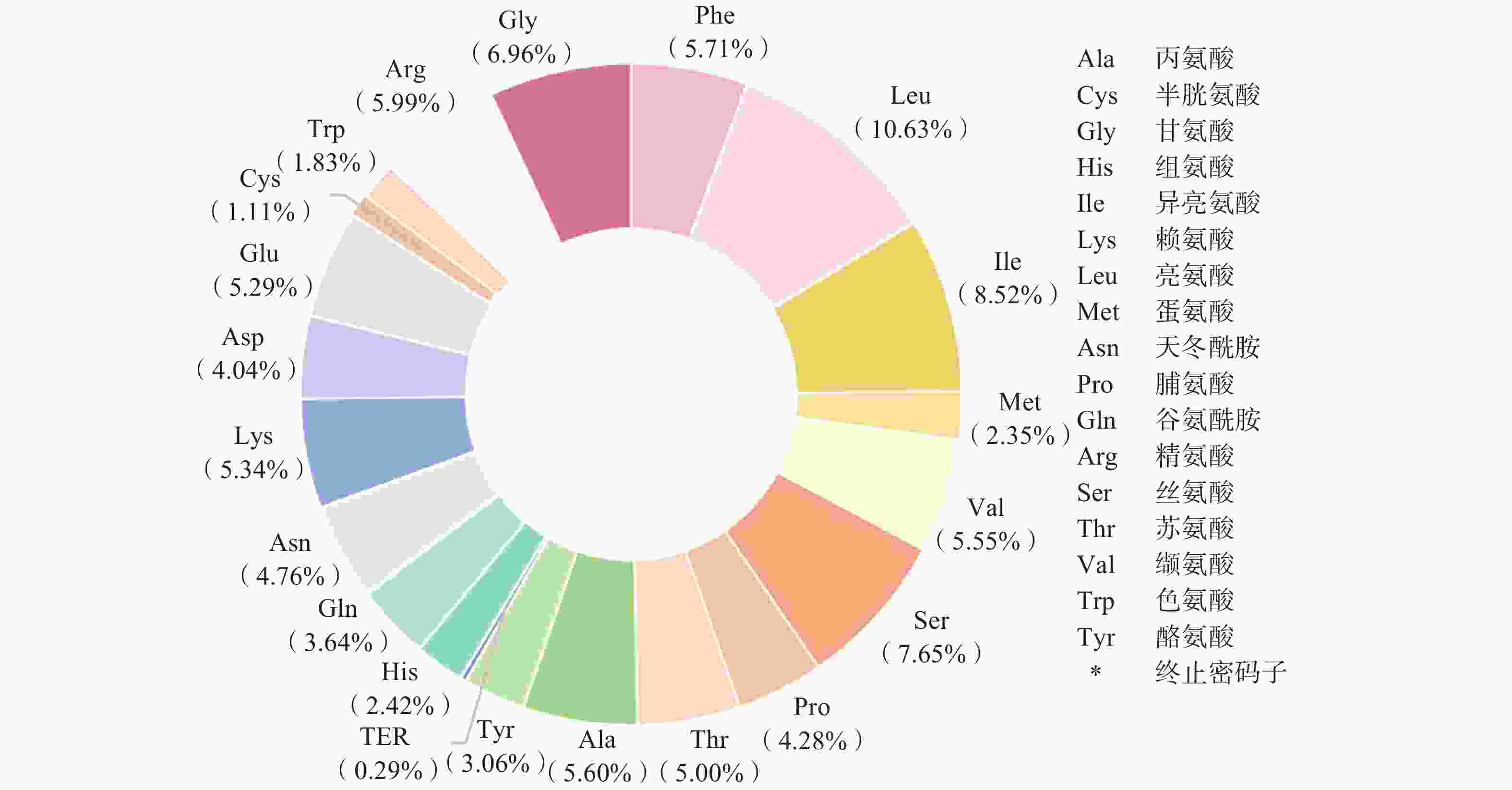
图 3 125个鼠尾草属植物叶绿体基因组中所有蛋白质编码基因中21个氨基酸和终止密码子的密码子含量
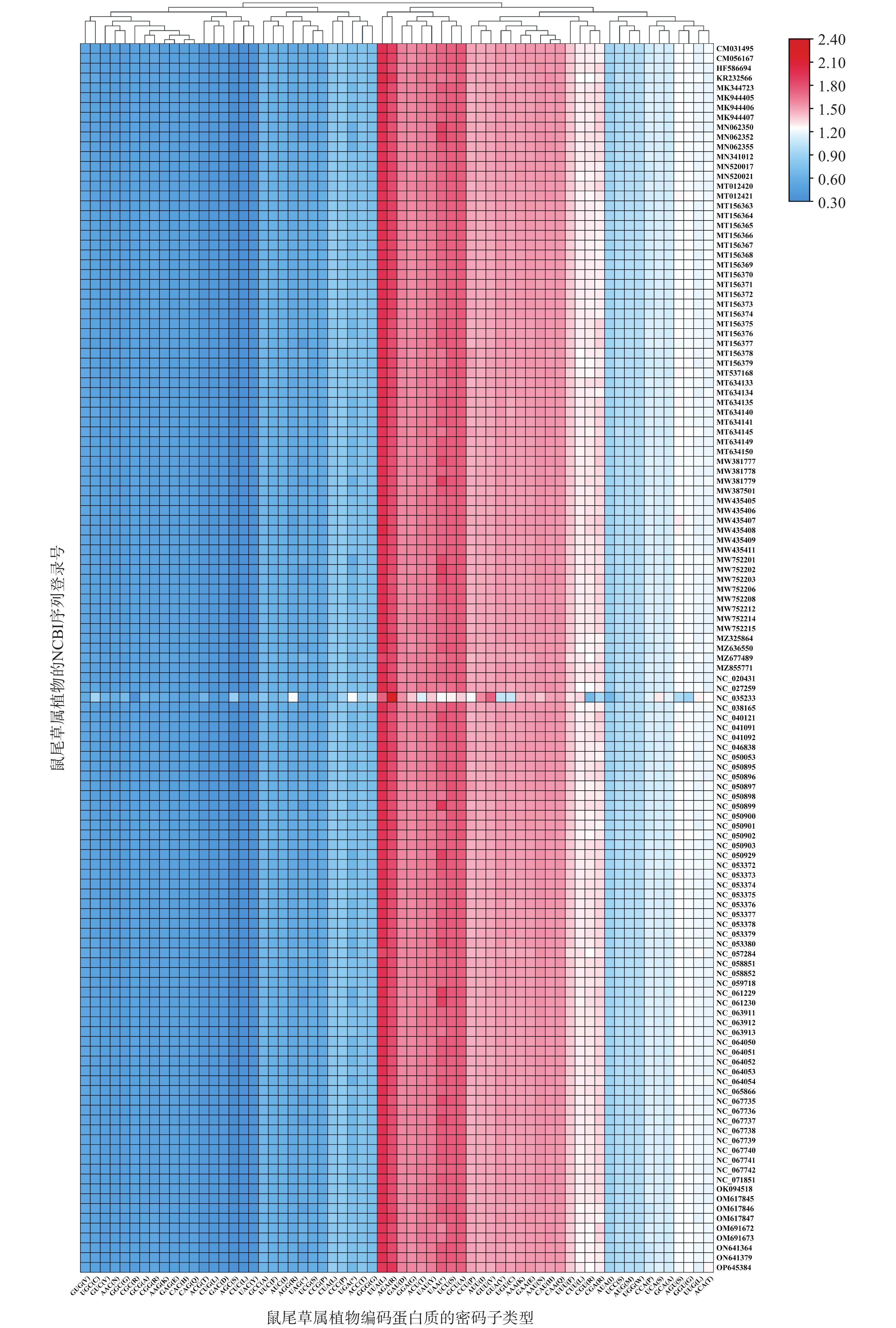
同义密码子相对使用度(RSCU)分析表明(图4),除Salvia japonica(NC_035233)外,其他鼠尾草属植物叶绿体基因组的RSCU表现出高度的相似性,在氨基酸中密码子大多以A/U碱基结尾,表现出明显的A/U偏好性。且UUA编码的亮氨酸整体为使用频率即RSCU最高的密码子,而AGC编码的丝氨酸为RSCU最低的密码子。
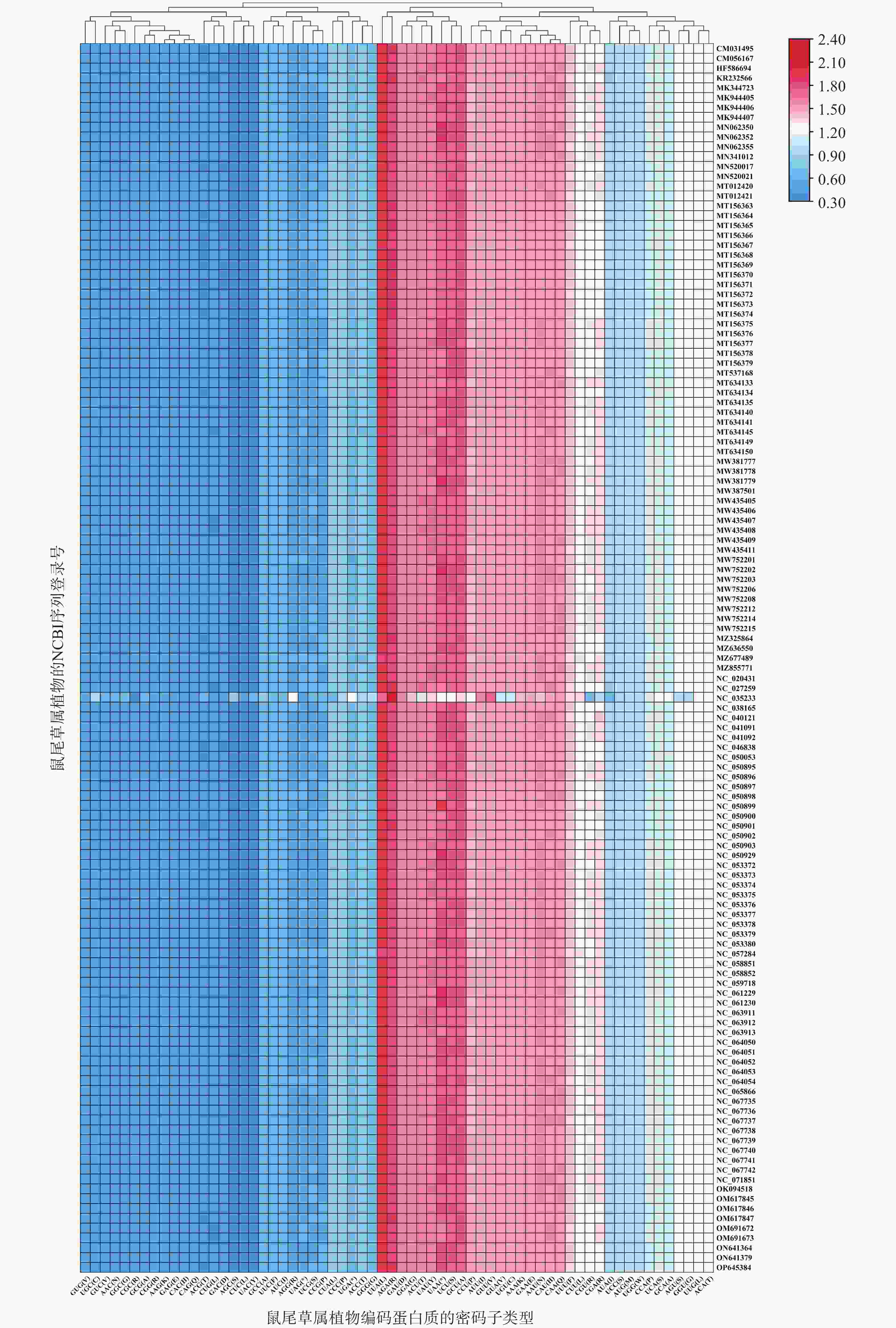
图 4 125个鼠尾草属植物叶绿体基因组相对同义密码子使用(RSCU)值的热图分析
-
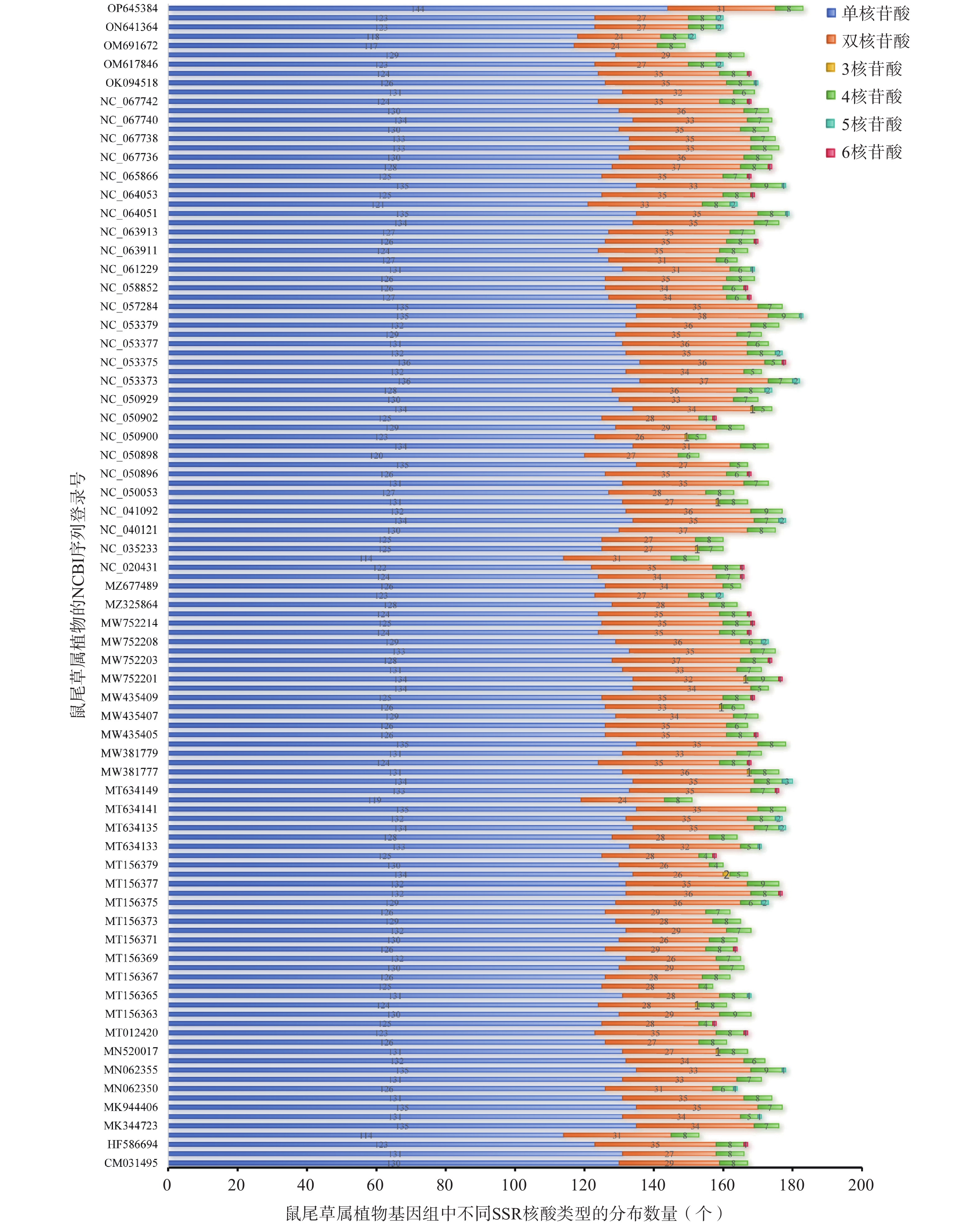
在125个鼠尾草属植物叶绿体基因组中,SSR的数量从149个(Salvia rosmarinus,OM691672)到183个(Salvia mekongensis,NC_053380;Salvia splendens,OP645384)不等(图5)。其中最常见的SSR类型是单核苷酸重复序列(占总SSR数量的76.24%),其次是双核苷酸重复序列(占总SSR数量的19.11%)、4核苷酸重复序列(占总SSR数量的4.27%)和5核苷酸重复序列(占总SSR数量的0.19%);3核苷酸重复序列所占比例最少(仅占总SSR数量的0.05%)。
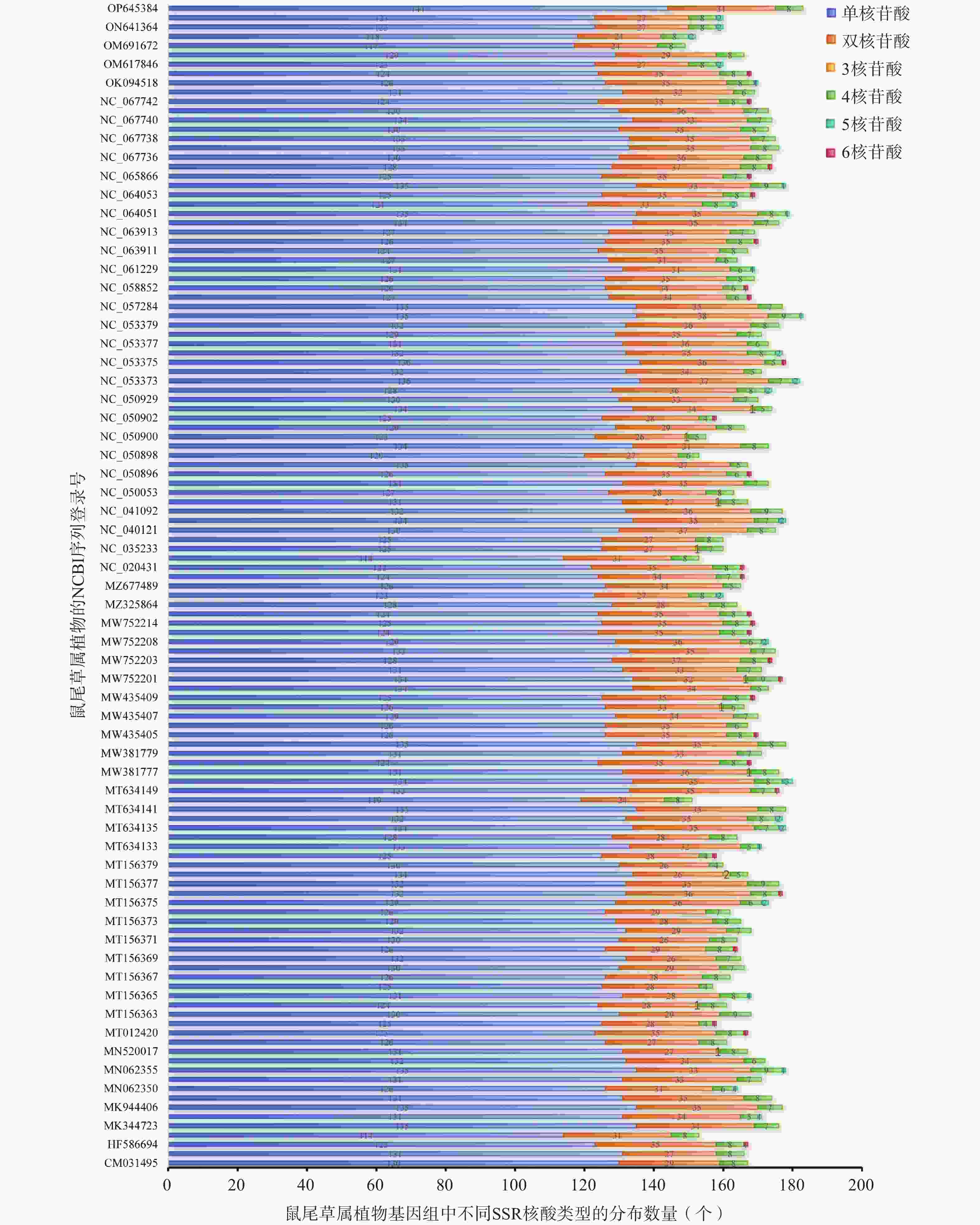
图 5 125个鼠尾草属植物叶绿体基因组中SSR不同核酸类型数量分布图
-
使用mVISTA软件以药用植物丹参叶绿体基因组为参考序列对125个鼠尾草属植物叶绿体基因组进行可视化分析表明,除了基因组的大小和基因定位略有变化外,基因的排列顺序和方向高度保守,并且没有观察到基因重排现象。此外,本研究发现编码区相对于非编码区更加保守,反向重复区比单拷贝区更保守。各基因位于trnQ-UUG-psbK、trnD-GUC-psbM、psaA-ycf3、cemA-ycf4和trnL-UAF-rpl32等的基因间隔区即非编码区,表现出较大的差异。在编码区中,matK、accD、ndhF和ycf1基因序列相似度较低。本研究还发现,S. glabrescens和S. chanryoenica植物中的ndhK基因及附近部分基因与药用植物丹参存在明显的变异,S. prewalskii、S. sp.HW-2020和S. digitaloides植物中的ndhB基因及附近部分基因和S. nanchuanensis植物中的rpl23基因及附近部分基因也发生了类似情况。
-
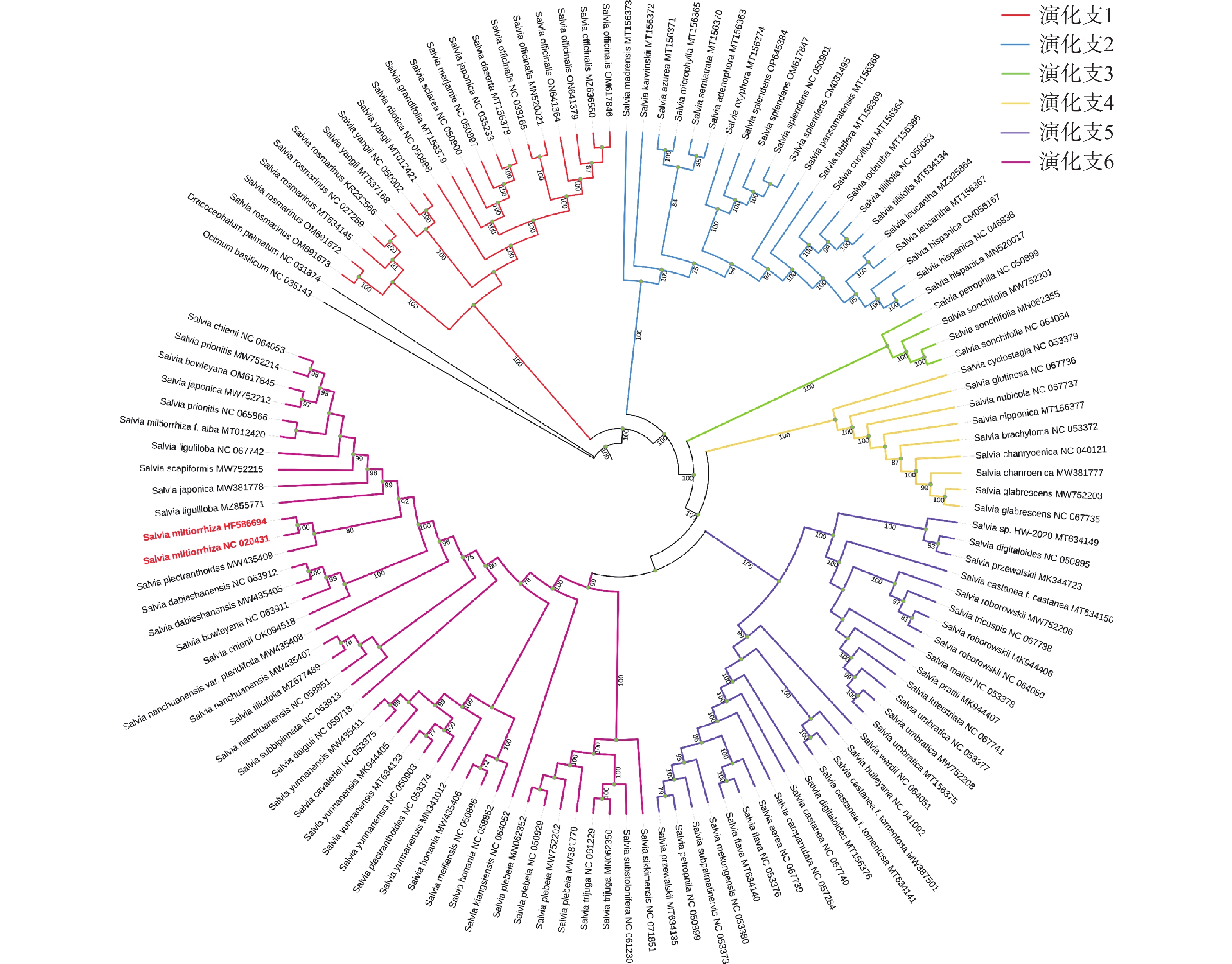
为了探究鼠尾草属植物的种间进化关系,本研究对整理的125个鼠尾草属植物进行了系统发育分析。以唇形科青兰属植物羽叶枝子花(Dracocephalum palmatum)和唇形科罗勒属植物罗勒(Ocimum basilicum)作为外类群,通过最大似然法,以叶绿体全基因组共有基因构建系统进化树,采用IQtree筛选出的最优TVM+F+I+G4核苷酸替换模型。结果显示(图6),125个鼠尾草属植物主要被分为6个进化分支,用不同颜色进行区分,药用植物丹参被聚类到Clade Ⅵ分支且与长冠鼠尾草(Salvia plectranthoides)亲缘关系最近。此外,进化树分支中的同一物种不同个体大都聚类到一起,但也出现了同一物种不同个体被分开的情况。
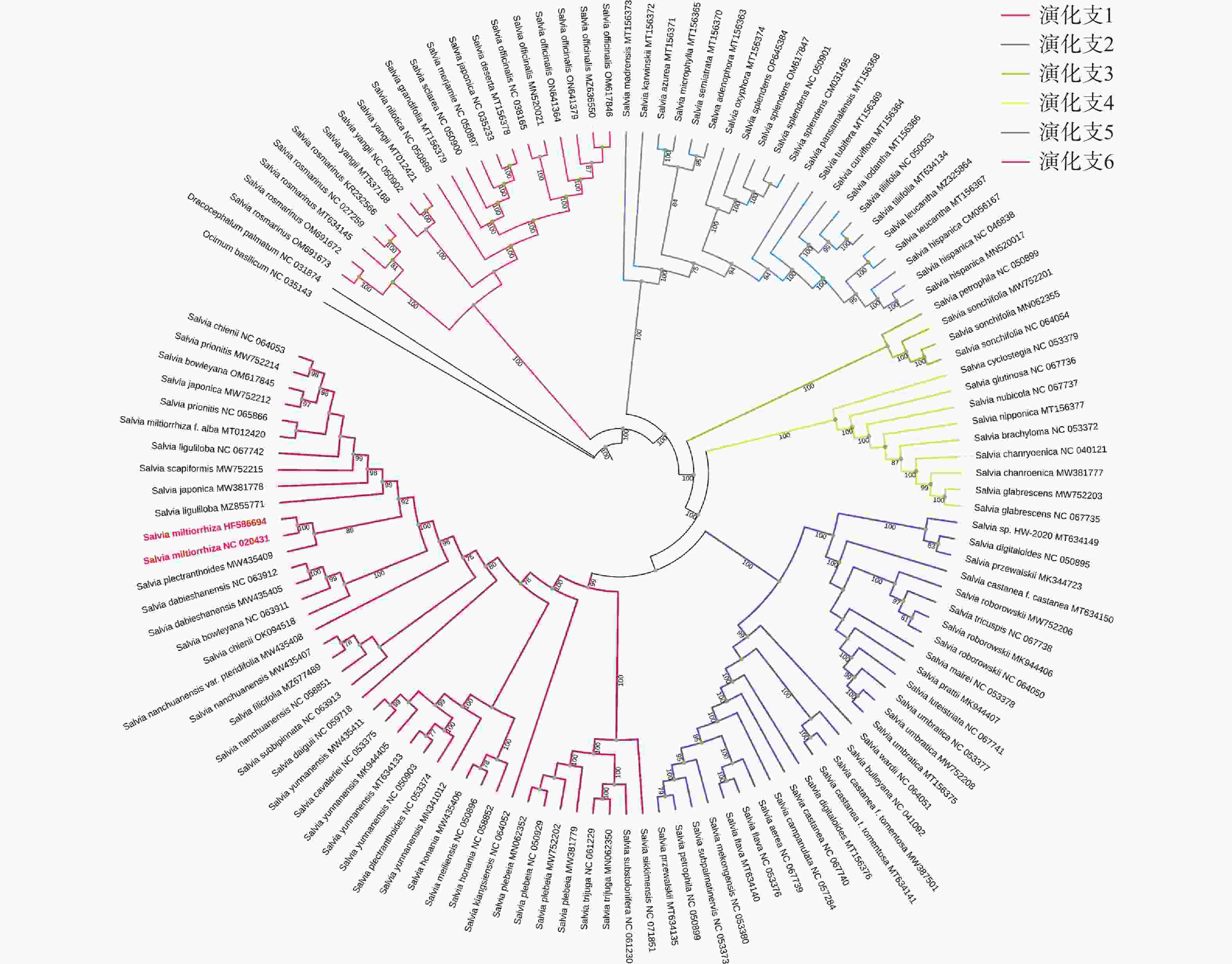
图 6 125个鼠尾草属植物的系统进化树(ML)
-
自第1条完整的烟草叶绿体基因组公布以来[12],叶绿体基因组研究得到了广泛关注,大量叶绿体基因组被公布[13]。叶绿体基因组的获得为植物亲缘关系鉴定和叶绿体转化技术研究提供了有力支撑[14]。叶绿体基因组的结构信息是实现外援基因精准导入,插入位点选择和叶绿体基因组改造的基础。叶绿体基因组数据匮乏是阻碍叶绿体代谢工程研究的一个主要限制。长期以来,一直认为叶绿体基因组序列是高度保守的。然而,只有约50%的叶绿体基因组含有高度保守的编码序列,对外源基因整合至关重要或含有调控序列的基因间序列并不保守[15],如单子叶植物中没有保守的间隔区[16],双子叶植物茄科物种中,多于150个基因间序列中只有4个是保守的。据报道,在表达盒的外源侧翼序列中的一个核苷酸取代也可能导致同源重组(HR)机制介导的整合频率的显著降低[17]。因此,了解叶绿体基因组在结构和序列方面的多样性,对于开发有效的基因代谢工程系统至关重要。
本研究通过重新注释分析125个鼠尾草属植物的叶绿体基因组,确保了注释结果的统一性和准确性,为该物种后续的叶绿体基因组相关分析工作提供了支持。鼠尾草属植物叶绿体基因组的密码子偏好性分析、SSR分析、叶绿体基因组结构的比较分析均表现出高度的相似性,这或与叶绿体基因组结构和编码区序列的高度保守有关。
此外,在鼠尾草属植物叶绿体基因组的注释分析过程中发现,部分鼠尾草属植物的叶绿体基因组出现了基因缺失现象。禾本科Bromus vulgaris植物的叶绿体基因组也存在基因缺失现象,缺少了部分tRNA基因[18]。tRNA基因缺失可能是由种间差异造成的,也可能是由于叶绿体基因组组装注释中出现的错误造成的[12],还需进一步探究。在叶绿体基因组中,有些基因由于缺乏开放阅读框架而被注释为假基因,如ycf基因。鼠李科(Rhamnaceae)植物叶绿体基因组中缺失了ycf15基因[19],本研究中Salvia grandifolia也缺失了ycf15基因,这可能与假基因有关。
在鼠尾草属植物叶绿体基因组结构的比较分析中发现,ndhF基因的序列相似度较低,属于高可变区。对于Salvia splendens和Salvia splendens植物中ndhF基因的缺失或与其位于高可变区有关。
125个(76种)鼠尾草属植物进化树分析表明,其主要被分为6个进化分支。结合已发表鼠尾草亚属分类[20]对进化树分支与亚属关系进行探究,发现位于CladeⅠ的Salvia yangii和Salvia rosmarinus分别属于Perovskia亚属和Rosmarinus亚属,在前人的研究中[20]被分为PR分支,而这2个鼠尾草属植物在进化树分支上均可进一步划分而独立于CladeⅠ分支,亦可将其看成PR分支。此外,位于CladeⅠ分支的鼠尾草属植物均属于Sclarea、Salvia、Heterosphace亚属,这与利用鼠尾草属植物全基因组所建进化树分支亚属种类保持一致[20]。CladeⅡ分支的鼠尾草属植物均属于Calosphace亚属,这一结果从侧面印证了此进化树关系分支的准确性。在前人研究的进化树分支中[20],进化树只分了4支,分别为 PR分支、CladeⅠ分支、CladeⅡ分支和CladeⅣ分支。PR分支、CladeⅠ分支和CladeⅡ分支同本研究进化树分支高度一致,CladeⅣ分支均属于Glutinaria亚属[19]。本研究CladeⅣ分支、CladeV分支和CladeⅥ分支大多也同样为Glutinaria亚属,与CladeⅣ分支[20]基本一致。此外,本研究还发现Salvia亚属在CladeⅠ、CladeⅢ和CladeⅣ中均有分布,此现象还需进一步分析。
叶绿体基因组作为植物基因组学的关键组成部分,在揭示叶绿体DNA的结构与起源、植物分子标记、分子育种以及遗传转化和叶绿体基因工程等方面具有重要科研价值。本研究对丹参及其同属共76种植物的125个叶绿体基因组进行了全面分析和比较,包括序列特征、密码子偏好性、SSR、叶绿体基因组边界收缩扩张及系统进化分析,提高了对丹参及其同属共76种植物叶绿体基因组的认知,为后续该属植物的叶绿体基因工程、遗传多样性分析、分子育种及物种鉴定等研究奠定了基础。
Analysis of chloroplast genomes from Salvia miltiorrhiza and its congeneric species
-
摘要: 丹参为唇形科鼠尾草属植物,是中医临床治疗心脑血管疾病的常用药物之一。叶绿体是植物细胞中由双层膜构成的含有叶绿素并能进行光合作用的一类细胞器。叶绿体基因组的结构信息,是实现外援基因精准导入、插入位点选择和叶绿体基因组改造的基础。该研究对丹参及其同属共76种植物的125个叶绿体基因组进行了全面分析和比较,包括序列特征、密码子偏好性、简单重复序列(SSR)、叶绿体基因组边界收缩扩张及系统进化分析,为后续该属植物的叶绿体基因工程、遗传多样性分析、分子育种及物种鉴定等研究提供了理论依据。Abstract: Salvia miltiorrhiza Bunge (Lamiaceae) is a medicinal plant widely used in Traditional Chinese Medicine for treating cardiovascular and cerebrovascular diseases. Chloroplasts are double-membrane-bound, chlorophyll-containing organelles and responsible for photosynthesis in plant cells. The structural information of chloroplast genomes serves as the foundation for precise exogenous gene insertion, site selection, and chloroplast genome modification. In this study, a comprehensive analysis and comparison of 125 chloroplast genomes from S. miltiorrhiza and 76 congeneric species were conducted, focusing on sequence characteristics, codon usage bias, simple sequence repeats (SSRs), contraction/expansion of chloroplast genome boundaries, and phylogenetic relationships, which could provide a theoretical foundation for advancing chloroplast genetic engineering, genetic diversity analysis, molecular breeding, and species identification within the Salvia genus.
-
图 1 鼠尾草属植物叶绿体基因组示意图
注:彩色图例表示不同功能的基因分组情况,图外圆圈黑色粗线表示反向重复序列区域(IRa和IRb)的范围,它们将基因组分为小单拷贝区域(SSC)和大单拷贝区域(LSC);圆圈内侧的基因顺时针转录,圆圈外侧的基因逆时针转录;深灰色的内圈对应GC含量,浅灰色对应AT含量。
-
[1] FUENTES P, ARMAREGO-MARRIOTT T, BOCK R. Plastid transformation and its application in metabolic engineering[J]. Curr Opin Biotechnol, 2018, 49:10-15. doi: 10.1016/j.copbio.2017.07.004 [2] CORRIVEAU J L, COLEMAN A W. Rapid screening method to detect potential biparental inheritance of plastid DNA and results for over 200 angiosperm species[J]. American J Botany, 1988, 75(10):1443-1458. doi: 10.1002/j.1537-2197.1988.tb11219.x [3] BHARADWAJ R, KUMAR SR, SATHISHKUMAR R. Green biotechnology: a brief update on plastid genome engineering[J]. Adv Plant Transgenics Methods Appl, 2019,20: 79-100. [4] RODRÍGUEZ-EZPELETA N, BRINKMANN H, BUREY S C, et al. Monophyly of primary photosynthetic eukaryotes: green plants, red algae, and glaucophytes[J]. Curr Biol, 2005, 15(14):1325-1330. doi: 10.1016/j.cub.2005.06.040 [5] ZHOU J G, CHEN X L, CUI Y X, et al. Molecular structure and phylogenetic analyses of complete chloroplast genomes of two Aristolochia medicinal species[J]. Int J Mol Sci, 2017, 18(9):1839. doi: 10.3390/ijms18091839 [6] ALTSCHUL S F, GISH W, MILLER W, et al. Basic local alignment search tool[J]. J Mol Biol, 1990, 215(3):403-410. doi: 10.1016/S0022-2836(05)80360-2 [7] SHI L C, CHEN H M, JIANG M, et al. CPGAVAS2, an integrated plastome sequence annotator and analyzer[J]. Nucleic Acids Res, 2019, 47(W1):W65-W73. doi: 10.1093/nar/gkz345 [8] DUNN N A, UNNI D R, DIESH C, et al. Apollo: democratizing genome annotation[J]. PLoS Comput Biol, 2019, 15(2):e1006790. doi: 10.1371/journal.pcbi.1006790 [9] FRAZER K A, PACHTER L, POLIAKOV A, et al. VISTA: computational tools for comparative genomics[J]. Nucleic Acids Res, 2004, 32(Web Server issue): W273-W279. [10] NGUYEN L T, SCHMIDT H A, VON HAESELER A, et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies[J]. Mol Biol Evol, 2015, 32(1):268-274. doi: 10.1093/molbev/msu300 [11] XIONG B, WANG T, HUANG S J, et al. Analysis of Codon usage bias in xyloglucan endotransglycosylase(XET)genes[J]. Int J Mol Sci, 2023, 24(7):6108. doi: 10.3390/ijms24076108 [12] SHINOZAKI K, OHME M, TANAKA M, et al. The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression[J]. EMBO J, 1986, 5(9):2043-2049. doi: 10.1002/j.1460-2075.1986.tb04464.x [13] ZHU T, ZHANG L, CHEN W, et al. Analysis of chloroplast genomes in 1 342 plants[J]. Genomics and Applied Biology, 2017, 36(10):4323-4333. [14] HE Y, HAN L, LIU Y, et al. Complete sequence analysis of chloroplast genome of salvia japonica[J]. Bulletin of Botanical Research, 2017, 37(4):572-578. [15] DANIELL H, LEE S B, GREVICH J, et al. Complete chloroplast genome sequences of Solanum bulbocastanum, Solanum lycopersicum and comparative analyses with other Solanaceae genomes[J]. Theor Appl Genet, 2006, 112(8):1503-1518. doi: 10.1007/s00122-006-0254-x [16] SASKI C, LEE S B, FJELLHEIM S, et al. Complete chloroplast genome sequences of Hordeum vulgare, Sorghum bicolor and Agrostis stolonifera, and comparative analyses with other grass genomes[J]. Theor Appl Genet, 2007, 115(4):571-590. doi: 10.1007/s00122-007-0567-4 [17] DANIELL H, LIN C S, YU M, et al. Chloroplast genomes: diversity, evolution, and applications in genetic engineering[J]. Genome Biol, 2016, 17(1):134. doi: 10.1186/s13059-016-1004-2 [18] REN J, TIAN J, JIANG H, et al. Comparative and phylogenetic analysis based on the chloroplast genome of Coleanthus subtilis (tratt.) seidel, a protected rare species of monotypic genus[J]. Front Plant Sci, 2022, 13:828467. doi: 10.3389/fpls.2022.828467 [19] WANICHTHANARAK K, NOOKAEW I, PASOOKHUSH P, et al. Revisiting chloroplast genomic landscape and annotation towards comparative chloroplast genomes of Rhamnaceae[J]. BMC Plant Biol, 2023, 23(1):59. doi: 10.1186/s12870-023-04074-5 [20] HU J D, QIU S, WANG F Y, et al. Functional divergence of CYP76AKs shapes the chemodiversity of abietane-type diterpenoids in genus Salvia[J]. Nat Commun, 2023, 14(1):4696. doi: 10.1038/s41467-023-40401-y -

 点击查看大图
点击查看大图
图(6)
计量
- 文章访问数: 4587
- HTML全文浏览量: 2417
- PDF下载量: 21
- 被引次数: 0
