

 下载:
下载:

-
仑伐替尼是一种新型的分子靶向药物,其打破了近10年来索拉非尼作为晚期肝细胞癌(HCC)一线治疗药物的垄断局势,为HCC治疗提供了新的策略[1]。仑伐替尼是一种多靶点酪氨酸激酶抑制剂(TKI),能够阻断血管内皮生长因子受体家族、成纤维细胞生长因子受体家族、血小板源性生长因子受体、酪氨酸激酶受体等的活性[2-6]。已有研究表明,仑伐替尼在抗肿瘤疗效上优于索拉非尼[7-8]。然而,仑伐替尼的水溶性差和生物利用度低,导致其在临床上需要增大剂量以发挥抗肿瘤作用,从而可能引起腹泻、厌食、呕吐、消化道出血,乃至胃肠道穿孔和形成胃肠瘘[9-11]。这些副作用限制了仑伐替尼在HCC治疗中的广泛应用。因此,通过制剂技术改善仑伐替尼的溶解性,降低其不良反应,增强其治疗效果,成为仑伐替尼的研究重点。既往研究显示纳米载体(如胶束、混合胶束、聚合物、脂质体和纳米颗粒等)已被用于改善药物的溶解度和递送效果[12-15]。
近年来,胶束在药物递送、基因传递和诊断成像等多个生物医学领域展现了重要作用。胶束主要由表面活性剂分子通过自发组装形成,具有疏水核心和亲水外壳的结构,能够包裹疏水药物,提高其在体内的溶解度和生物利用度[16-17]。此外,通过调节胶束的组成和结构,可以优化药物的释放特性[18]。而混合胶束利用不同嵌段共聚物的互补特性,具有克服单一聚合物胶束在载药量低和稳定性不足等方面局限的优势[19-21]。
本研究选用具有良好生物相容性、低毒性和增溶性的两亲性嵌段共聚物 P123和 F127作为载体材料,对肝癌一线治疗药物仑伐替尼进行包载。通过星点设计-效应面法优化仑伐替尼混合胶束(LFT-MMs)的工艺处方,为药物递送系统的进一步研究提供科学依据。
-
RV 10 D S96旋转蒸发仪(德国IKA);SHB-Ⅲ循环水式多用真空泵(郑州长城科工贸有限公司);Scientz-10N冷冻干燥机(宁波新芝生物科技股份有限公司);KMS-101B磁力搅拌器(上海精凿科技有限公司);Waters
2487 高效液相色谱仪(美国waters公司);N-2000色谱工作站数据处理系统(浙江大学);AE240电子天平、S210 pH计(梅德勒-托利多仪器有限公司);Mastersizer3000 马尔文粒度测定仪(英国Malvern Panalytical Ltd公司);K-82J真空干燥箱(上海博迅医疗生物仪器股份有限公司);JP-100超声波清洗机[深圳市结盟清洗设备(上海)有限公司]。 -
仑伐替尼(aladdin,纯度≥99%,批号:D2219078,购自阿拉丁试剂有限公司);P123(批号:10111562547)、F127(批号:101141111)均为德国 Sigma-Aldrich 公司;甘露醇(批号:20180528)、乙酸铵(分析纯,批号:20160118)均为天津市盛奥化学试剂有限公司;磷酸(批号:20101005,天津永晟精细化工有限公司);甲醇(色谱纯,批号:34860-4 L-R)、乙腈(色谱纯,批号:34851-4 L)均为Sigma-Aldrich;其余试剂均为分析纯;水为灭菌注射用水。
-
采用薄膜水化法制备仑伐替尼混合胶束(LFT-MMs)。精密称取处方量的仑伐替尼原料药及载体材料 P123 和 F127,将其置于 50 ml 圆底烧瓶中,加入甲醇10 ml并超声处理以促进其溶解。在一定温度下旋转蒸发除去有机溶剂,使其形成一层均匀药物薄膜后,置于真空干燥箱中干燥过夜以彻底去除残余溶剂。向干燥膜中加入适量的同温灭菌注射用水在一定温度下进行水化,制得载药胶束溶液,并将溶液冷却至室温。此溶液通过0.22 μm有机微孔滤膜过滤,将其移入透析袋(分子量截留限为8 000),使用去离子水透析以去除杂质及未被包封的药物,最终获得无色透明的LFT-MMs溶液。向LFT-MMs溶液中加入10%的甘露醇作为冻干保护剂,冷冻干燥过夜,最终得到白色固体形态的LFT-MMs粉末,冷冻保存备用。
-
以DiKMA-C18(型号4.6 mm×150 mm,5 μm)为色谱柱,0.1%磷酸水溶液(乙酸铵调节pH至4.5)-90%乙腈(体积比 60∶40)为流动相,检测波长为300 nm,柱温为30℃,流速为1.0 ml/min,进样量为20 μl。
-
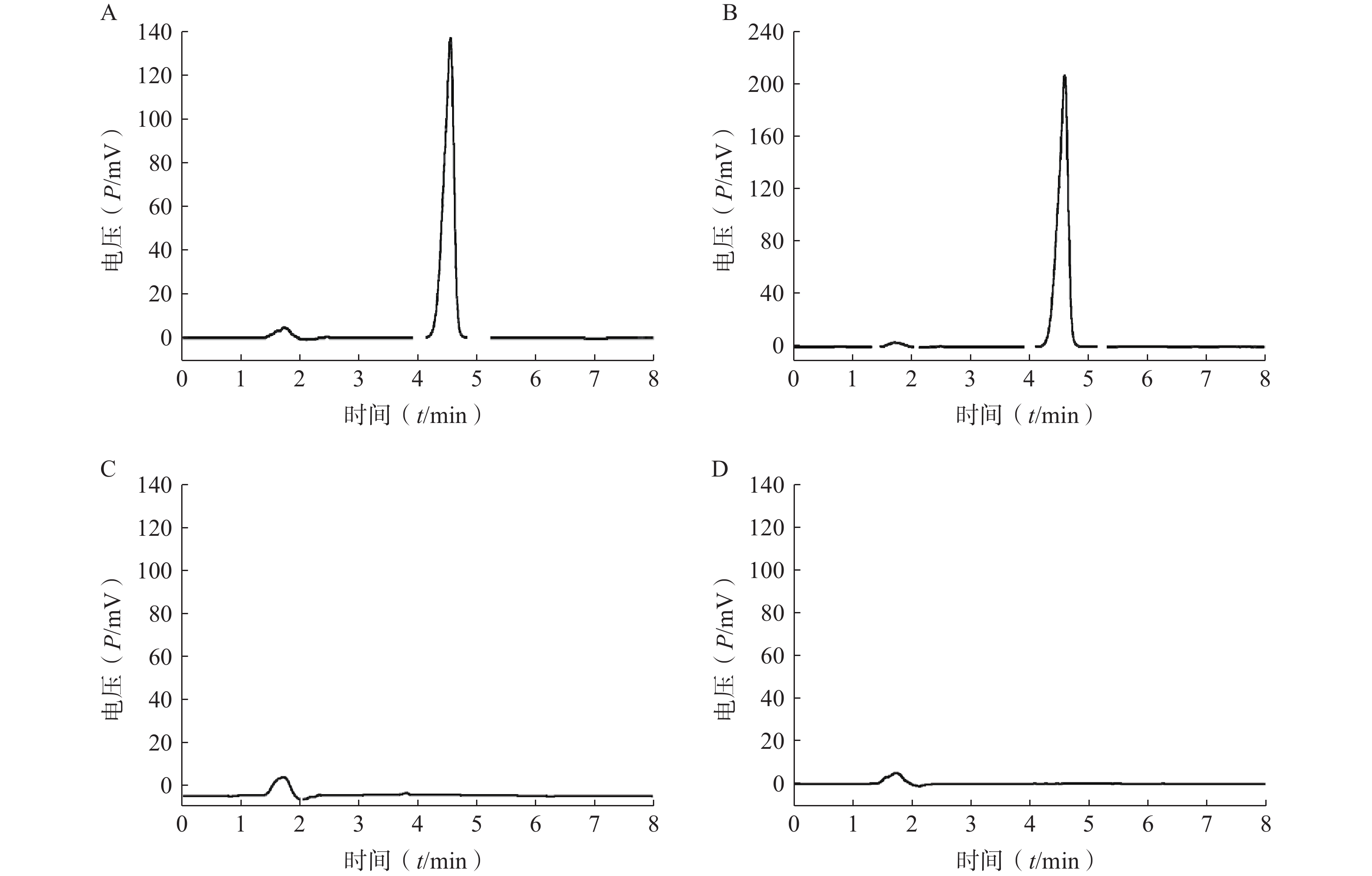
分取LFT-MMs溶液、空白胶束溶液、仑伐替尼对照品溶液,按“2.2.1”项条件进样分析,记录色谱图。如图1所示,仑伐替尼对照品的保留时间大约为 4.6 min, LFT-MMs溶液在相应的保留时间下有色谱峰出现,而空白胶束溶液在相应的时间下无色谱峰出现。该结果表明胶束的辅料P123和F127对仑伐替尼含量的测定无干扰,此方法专属性良好。
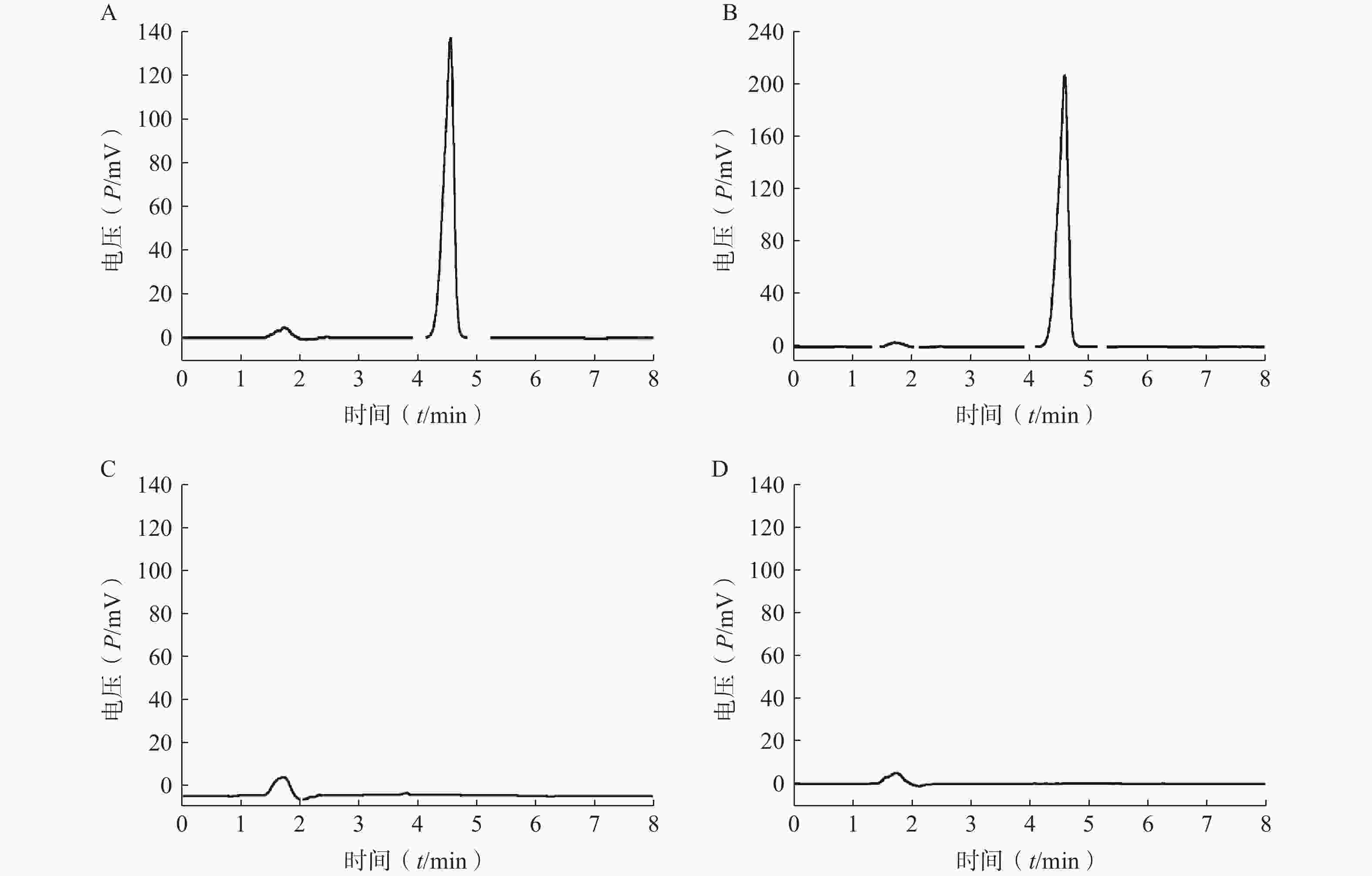
图 1 LFT-MMs的HPLC专属性谱图
-
准确称取10 mg仑伐替尼置于10 ml量瓶中,使用DMSO溶解,并通过0.22 μm微孔滤膜过滤后稀释至刻度,混匀,制备得到1 mg/ml的仑伐替尼对照品储备液。将储备液用甲醇逐步稀释,得到一系列质量浓度分别为100.0、80.0、60.0、40.0、20.0、10.0、5.0 μg/ml的对照品溶液。以仑伐替尼的质量浓度(C)作为横坐标,色谱峰面积(A)作为纵坐标,绘制标准曲线,并得到线性回归方程:A=23 514C+102 029,相关系数r= 0.999 9。结果显示,仑伐替尼的质量浓度在5.0~100.0 μg/ml范围内具有良好的线性关系。
-
在精密度实验中,低浓度(5 μg/ml)、中浓度(40 μg/ml)、高浓度(100 μg/ml)对照品溶液的日内精密度相对标准偏差(RSD)分别为0.73%、0.89%、0.40%,日间精密度RSD分别为1.02%、1.18%、0.87%(n=5)。在回收率测试中,低、中、高质量浓度的加标回收率分别为103.13%、99.93%、101.00%,RSD分别为0.35%、0.70%、0.25%(n=3)。经验证,所建立的检测方法准确且可靠。
-
取同LFT-MMs溶液,在 0、2、4、8、12、24 h,按“2.2.1”条件下测定仑伐替尼的峰面积值,计算 RSD 值为 1.83%,表明供试品溶液在 24 h内稳定性较好。
-
取适量制备的冻干胶束粉末用甲醇溶解,并稀释至一定浓度,超声破乳,经孔径 0.22 μm 微孔滤膜滤过,按“2.2.1”项下色谱条件进行定量。包封率(EE)和载药量(DL)的计算公式如下:
EE(%)=载药胶束中仑伐替尼含量 /仑伐替尼的投药量×100%;
DL(%)= 载药胶束中仑伐替尼含量/(仑伐替尼的投药量+载体材料的总质量)×100%。
-
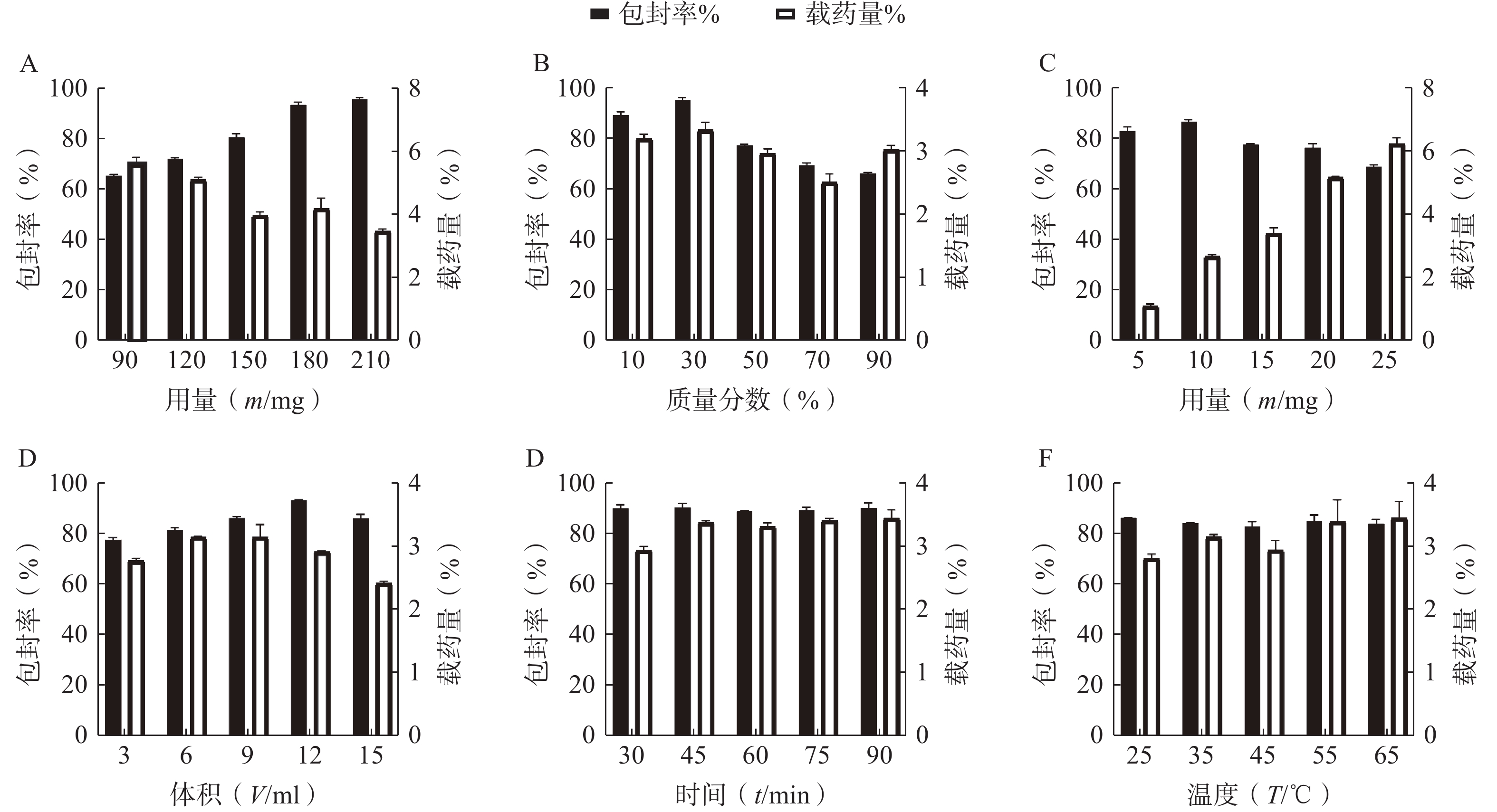
按“2.1”项下的制备方法,以 EE和DL为考察指标,通过单因素试验考察 P123 占载体材料的质量比(10%、30%、50%、70%和90%)、载体材料用量(90、120、150、180和210 mg)、水相体积(3、6、9、12和15 ml)、水化时间(30、45、60、75和90 min)、旋蒸温度(25℃、35℃、45℃、55℃和60℃)、有机溶剂用量(1、2、3、4和5 ml)的影响,结果见图2。根据单因素的初步考察,最终确定投药量为10 mg,旋蒸温度为 55℃,水化时间为45 min,而载体 P123 质量百分比、载体材料用量和水相体积对 EE和DL有显著影响,因此将它们作为待优化项进行星点设计-效应面法实验。

图 2 单因素考察(n=3,mean±SD)
-
在单因素试验的基础上,进一步采用星点设计-效应面法优化LFT-MMs制剂工艺。选择载体 P123 质量百分比(a)、P123-F127 混合载体用量(b)、水相体积(c)3 个显著因素作为考察因素,每因素设 5 个水平,因素水平见表1。应用 Design Expert 12软件以 EE(Y1) 和DL(Y2)为考察指标,设计共 20个试验点的效应面分析试验,见表2。
表 1 星点设计因素水平表
因素 P123质量百分比
(%)F127-P123用量
(m/mg)水化体积
(V/ml)−1.68 6.25 49.09 2.93 −1.00 25.00 90.00 6.00 0 52.50 150.00 10.50 1.00 80.00 210.00 15.00 1.68 98.75 250.91 18.07 表 2 星点设计表及效应值
序号 a:P123质量
百分比(%)b:载体材料
用量(m/mg)c:水化体积
(V/ml)包封率
(%)载药量
(%)1 80.00 90.00 6.00 90.79 9.53 2 52.50 150.00 10.50 80.95 5.95 3 52.50 150.00 10.50 77.21 6.19 4 25.00 90.00 6.00 59.43 4.91 5 52.50 150.00 10.50 79.97 6.16 6 98.75 150.00 10.50 90.04 5.62 7 80.00 210.00 6.00 77.86 6.96 8 25.00 90.00 15.00 43.77 4.98 9 80.00 210.00 15.00 94.30 4.10 10 52.50 150.00 10.50 84.36 6.82 11 6.25 150.00 10.50 79.19 6.46 12 25.00 210.00 6.00 85.31 3.62 13 52.50 150.00 18.07 76.50 4.33 14 52.50 150.00 10.50 83.22 6.96 15 52.50 250.90 10.50 80.85 3.33 16 52.50 150.00 10.50 82.82 6.99 17 80.00 90.00 15.00 68.94 7.95 18 25.00 210.00 15.00 83.54 5.29 19 52.50 49.09 10.50 52.12 8.99 20 52.50 150.00 2.93 70.97 8.09 -
分别对各因素各水平用Design Expert 12软件进行多元线性回归及模型拟合后,以模型回归项的差异显著(P<0.01)且R2是否接近1来评价模拟方程。发现以三次多项式拟合效果最好(R2接近1且P值较小),具体方程及评价指标如下:
Y1=81.42+5.43 a+6.71 b+2.77 c−18.82 ab+4.25 ac+18.45 bc+3.19 a2−12.28 b2−7.69 c2+14.51 abc−0.012 2 a2b−21.41 a2c+20.24 ab2 (r2=0.989 0,P=0.006 1);
Y2=6.51−0.42 a−1.402 b−1.88 c−1.92 ab−2.19 ac+0.113 2 bc+0.471 7 a2−0.907 3 b2−0.301 8 c2−1.71 abc−0.025 6 a2b+3.71 a2c+6.98 ab2 (r2=0.980 6,P=0.002 4)。
各方程的方差分析结果见表3。两个模型均具有显著性差异(P<0.05),能较好反映因素对效应值的影响。进一步分析各方程中的各项“a、b、ab、bc、b2 ”对包封率有显著性影响,发现a、b、a2、b2对载药量有显著性影响。
表 3 效应面拟合模型对包封率和载药量的方差分析
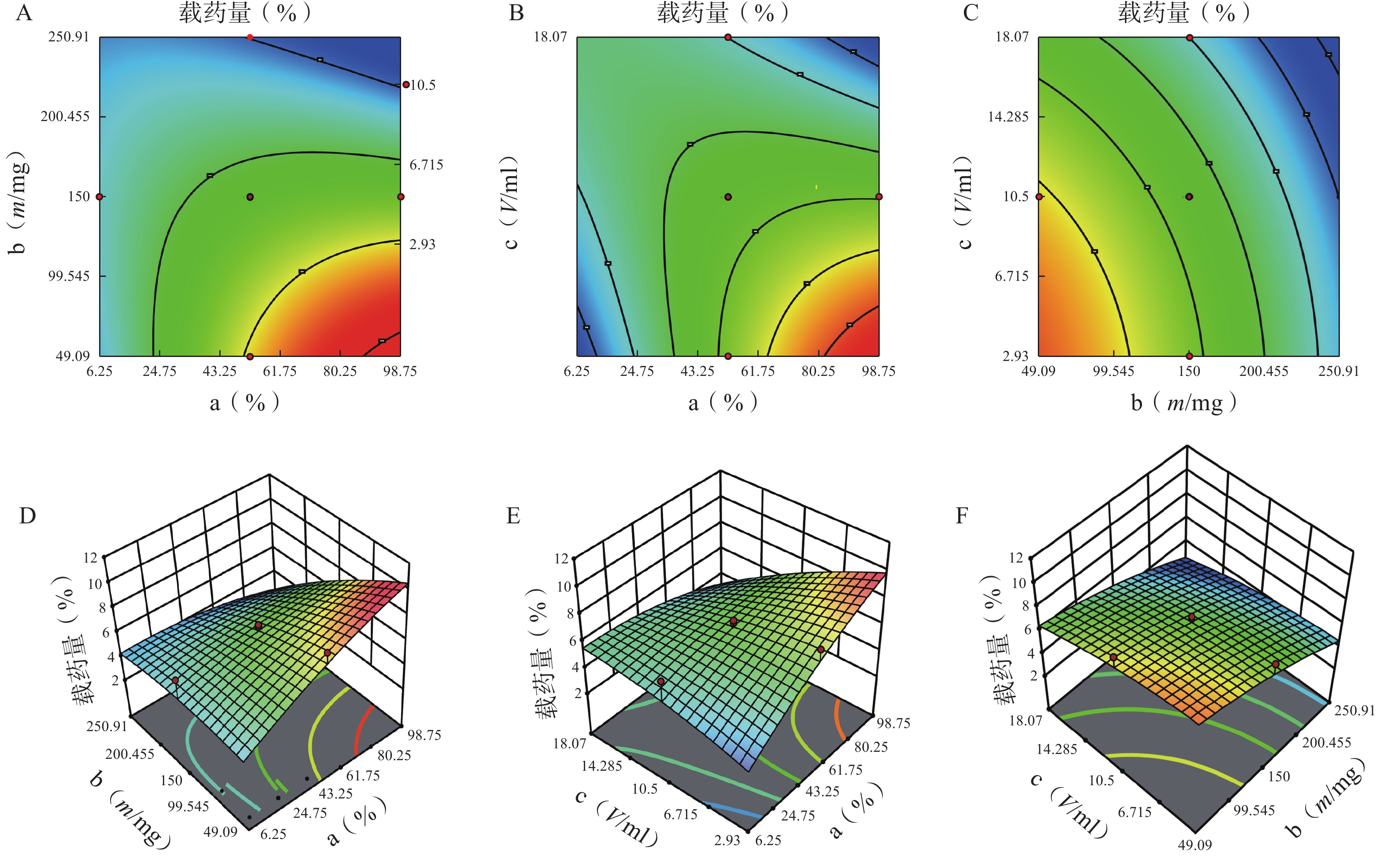
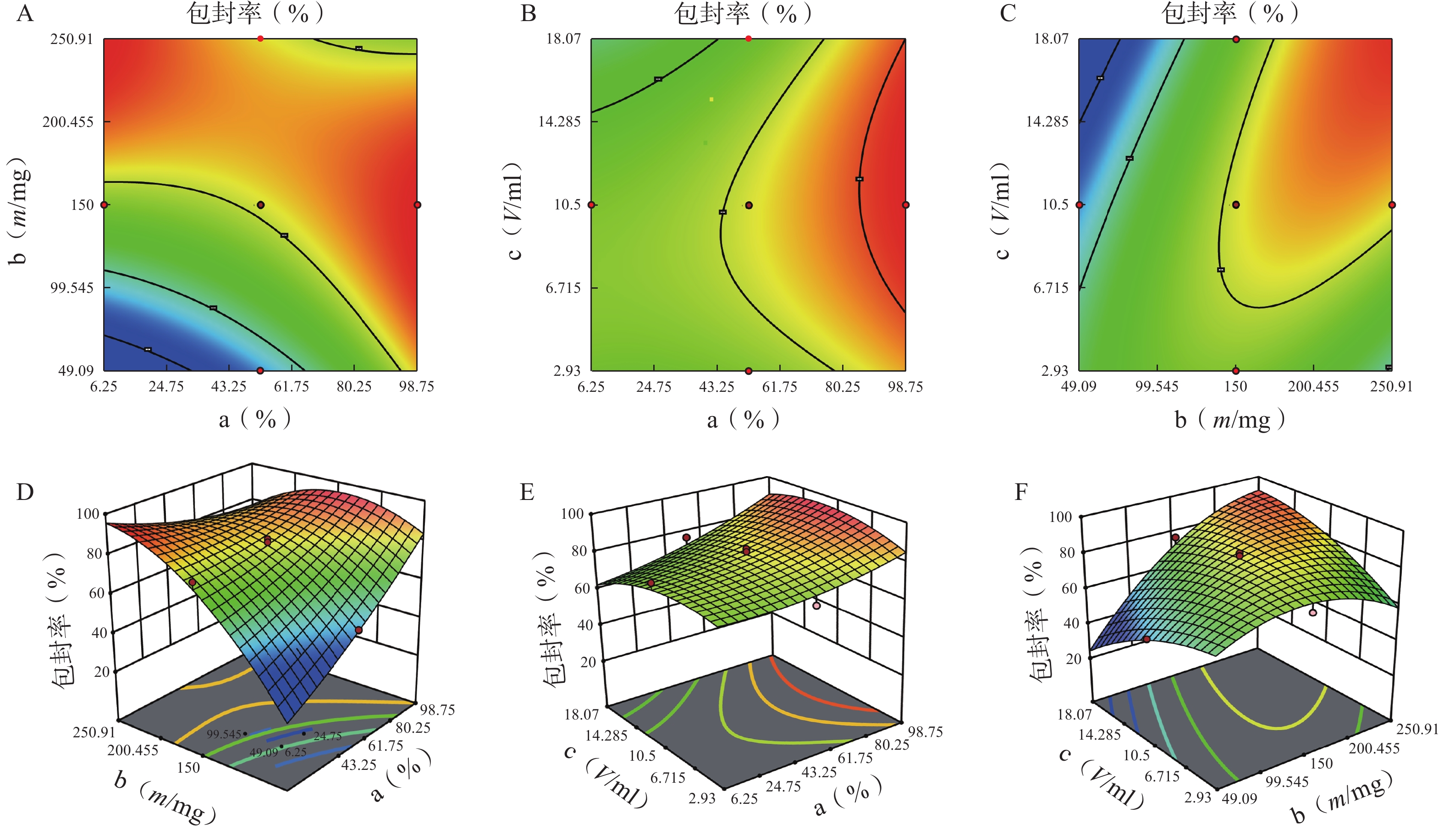
方差来源 自由度 包封率 载药量 平方和 均方 F值 P值 平方和 均方 F值 P值 模型 9 2833.07 314.79 12.95 <0.01 41.61 4.62 3.48 0.03 a 1 446.49 446.49 18.37 <0.01 5.08 5.08 3.82 0.08 b 1 1169.85 1169.85 48.12 <0.01 20.96 20.96 15.78 0.00 c 1 13.42 13.42 0.55 0.47 5.96 5.96 4.49 0.06 ab 1 354.05 354.05 14.56 <0.01 3.70 3.70 2.79 0.13 ac 1 18.06 18.06 0.74 0.41 4.77 4.77 3.59 0.09 bc 1 340.34 340.34 14.00 <0.01 0.01 0.01 0.01 0.92 a² 1 25.39 25.39 1.04 0.33 0.62 0.62 0.47 0.01 b² 1 372.27 372.27 15.31 <0.01 0.40 0.40 0.30 0.01 c² 1 91.46 91.46 3.76 0.08 0.32 0.32 0.24 0.64 残差 10 243.11 24.31 13.28 1.33 失拟项 5 209.21 41.84 6.17 0.03 12.21 2.44 11.44 0.01 净误差 5 33.89 6.78 1.07 0.21 总离差 19 3076.18 54.89 由以上方程可知,三项式拟合相关系数 R2 值高于多元线性回归及二次项拟合,失拟值无显著性,表明建立的模型可靠,拟合情况良好,故选择三项式拟合模型为最终模型。据上述回归模型绘制三维效应面图和二维等高线图,结果见图3、图4。软件分析优化得到LFT-MMs 的最优制备工艺为:P123 质量百分比为 80%,载体材料用量为 90 mg,水化体积为 6 ml。
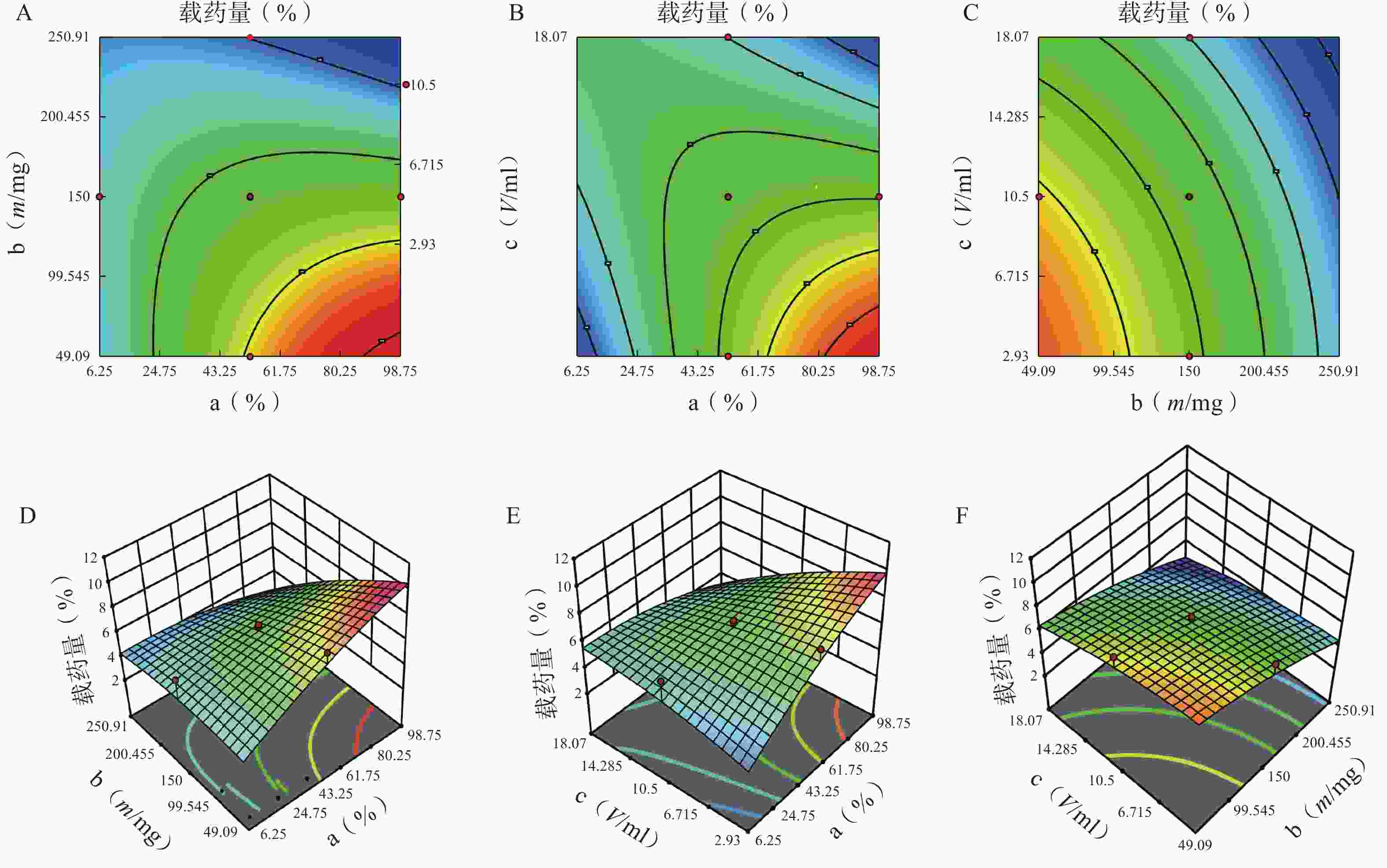
图 3 因素对载药量的等高线和效应面图
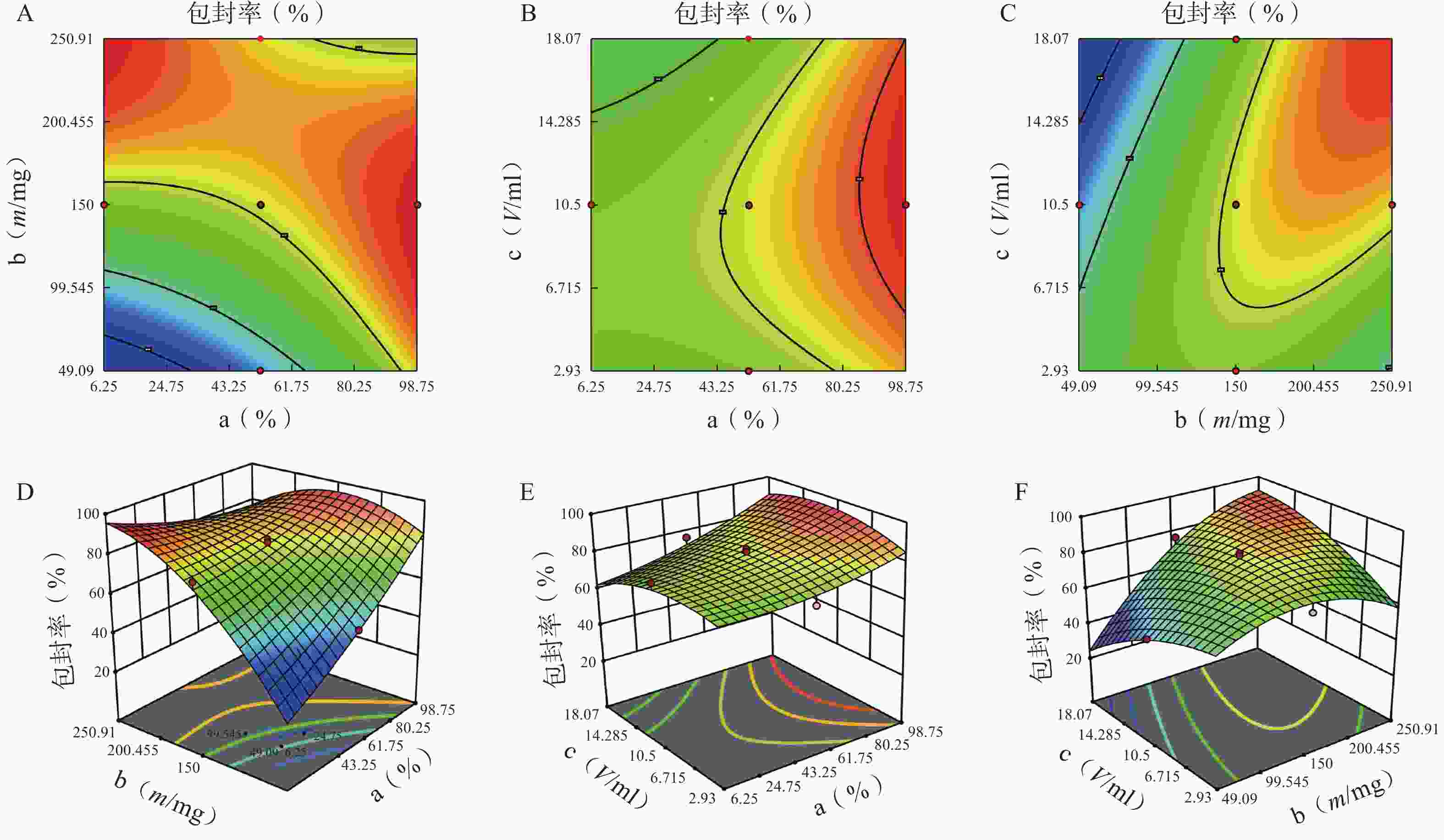
图 4 因素对包封率的等高线和效应面图
-
根据星点设计-效应面法得出的最佳处方工艺,制备 3批 LFT-MMs,并测量对每批胶束的EE和DL,取平均值与预测值进行比较。预测EE为84.24%,DL为8.67%,测得平均EE为(83.33±0.30)%,平均DL为(8.67±0.07)%。EE和DL预测值和实际测量值偏差较小且工艺重复性好,说明建立的三次回归模型,在试验范围内具有很好的预测性,可为载药胶束制备提供理论依据。
-
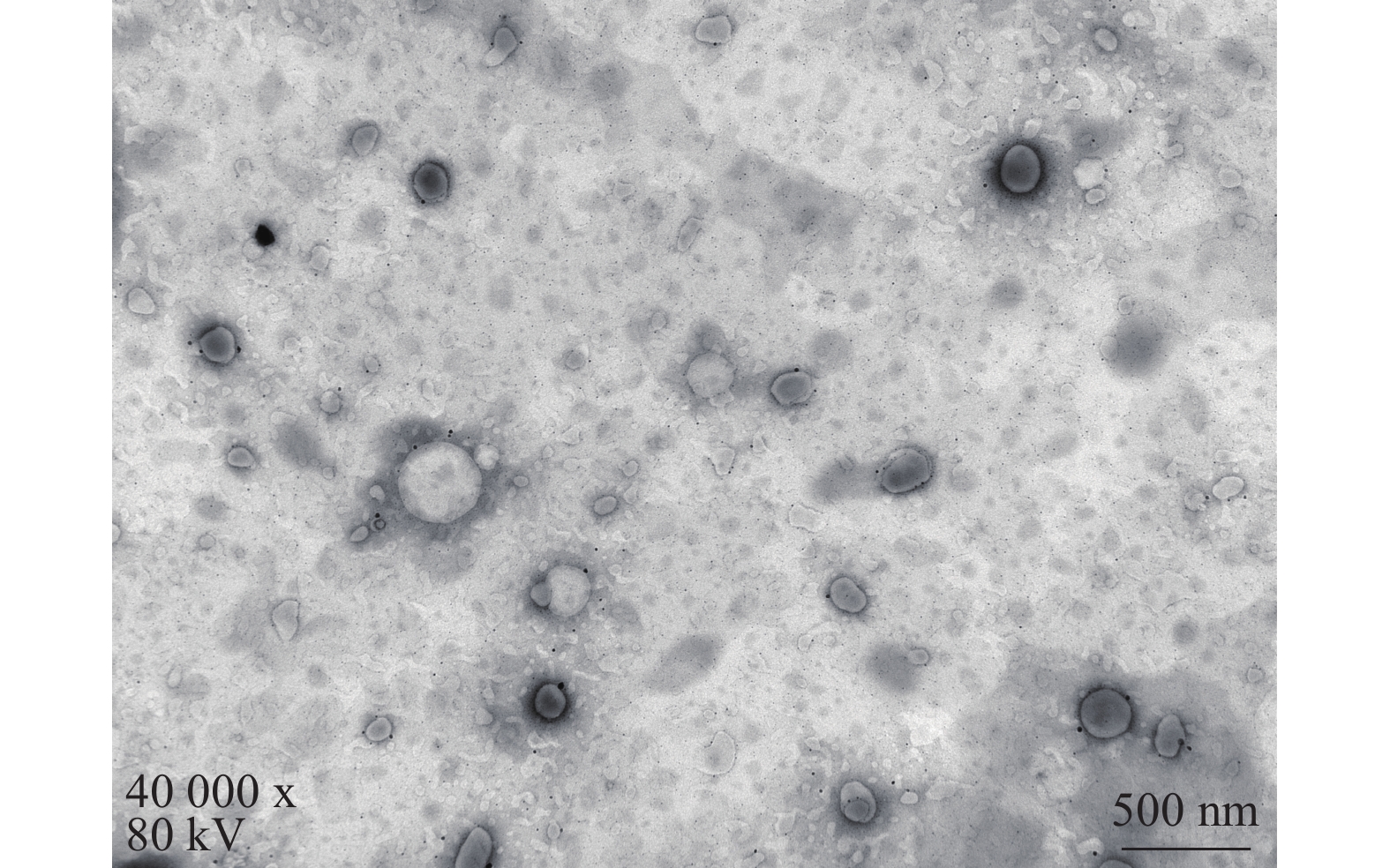
将最优工艺制备的 LFT-MMs冻干制剂用灭菌注射用水稀释到一定倍数后,取1滴于铜网上吸附后,用 2.0%磷钨酸负染,自然挥干,置透射电镜下观察微观形态并拍照,结果可见LFT-MMs的形态规整,呈圆球形,分散性好,无聚集(图5)。
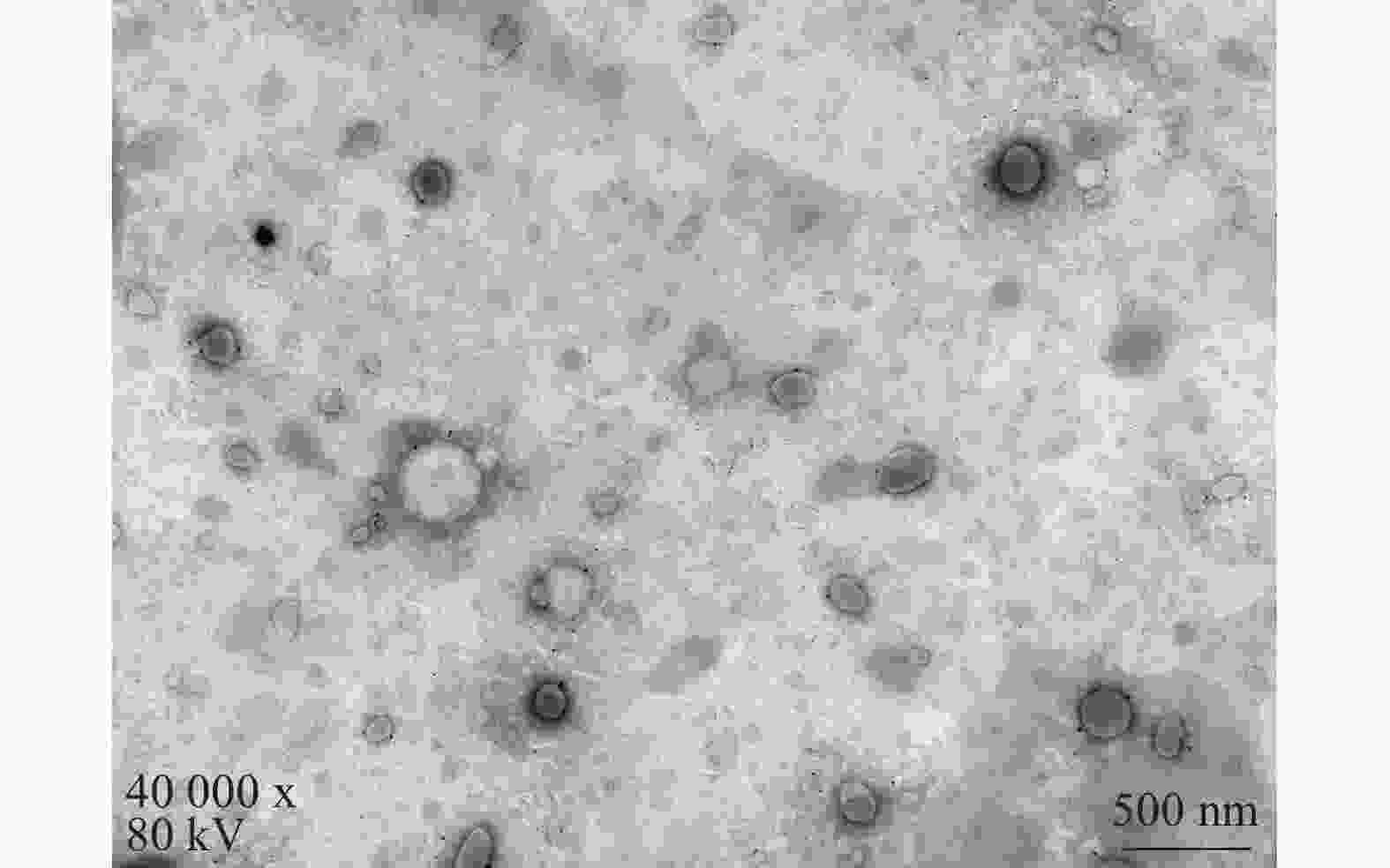
图 5 LFT-MMs透射电镜图(×40 000, 80 kV)
-
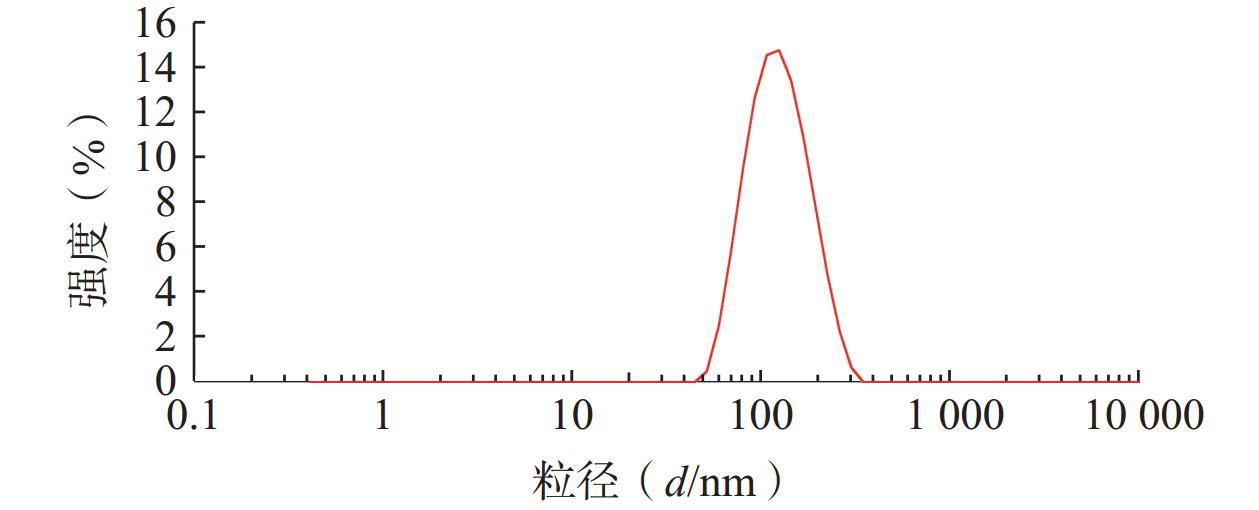
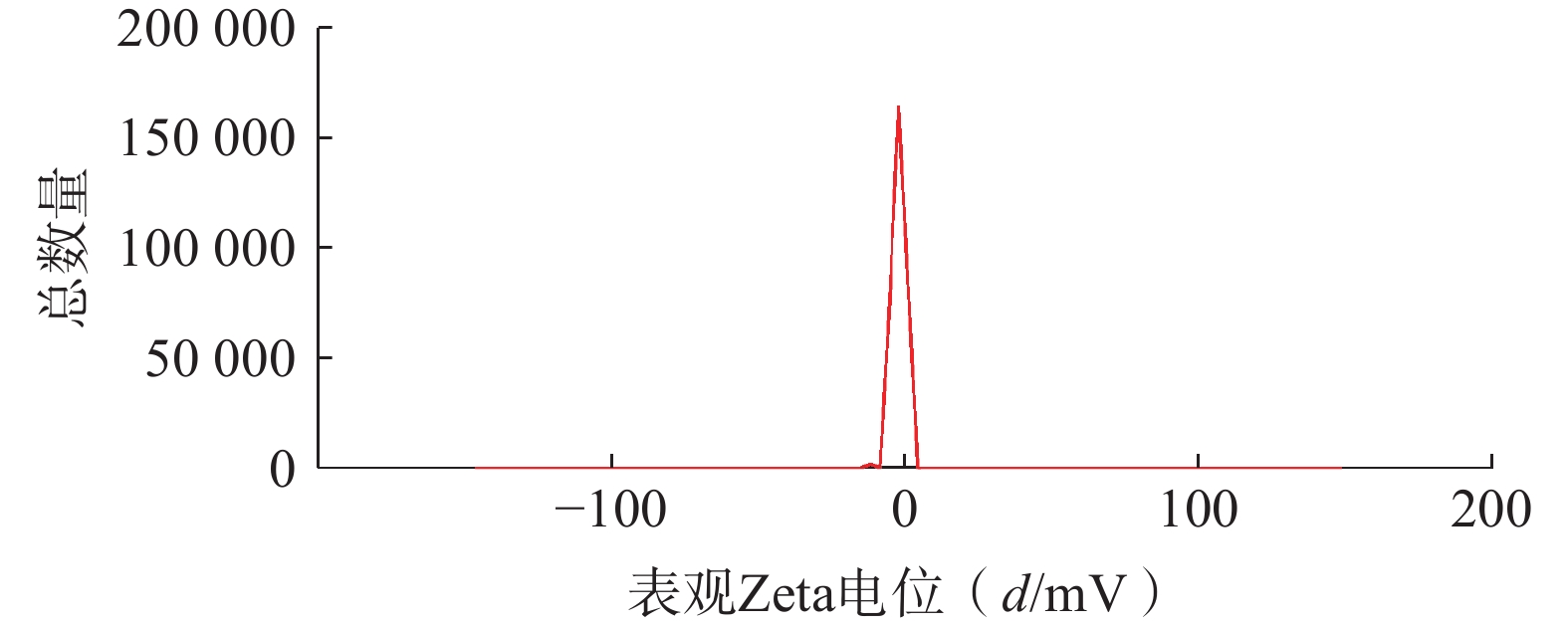
将最优工艺制备的 LFT-MMs冻干制剂用灭菌注射用水稀释到一定倍数后,使用马尔文纳米粒度电位仪测定胶束的粒径、 PDI及Zeta 电位,结果见图6、图7。LFT-MMs 的平均粒径、PDI 和 Zeta 电位分别为(104.0±0.32) nm、(0.22±1.19)和(−2.56±0.81) mV。
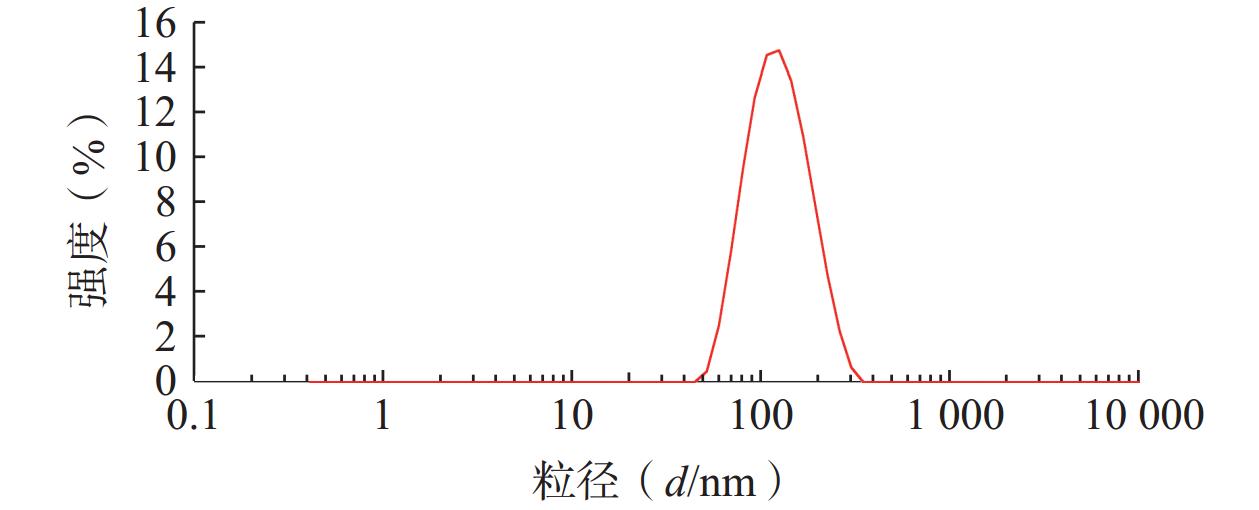
图 6 LFT-MMs 粒径分布图
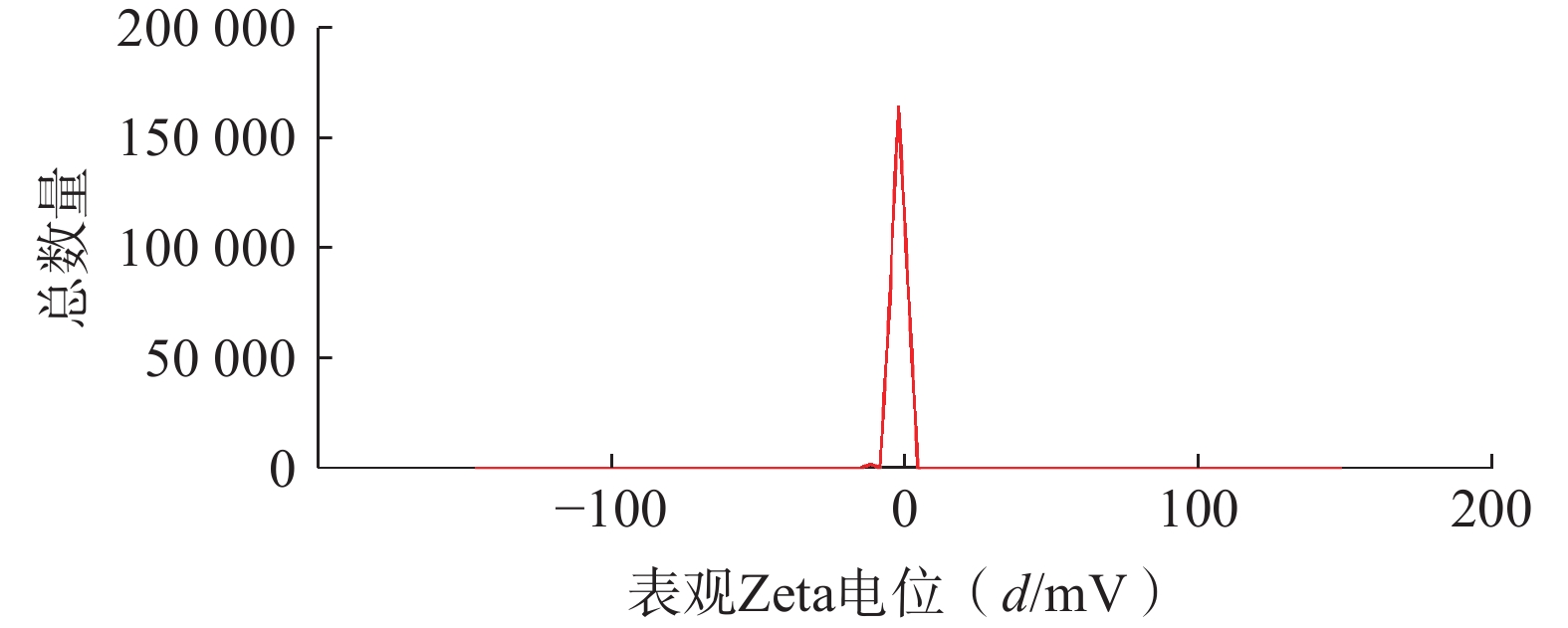
图 7 LFT-MMs Zeta电位分布图
-
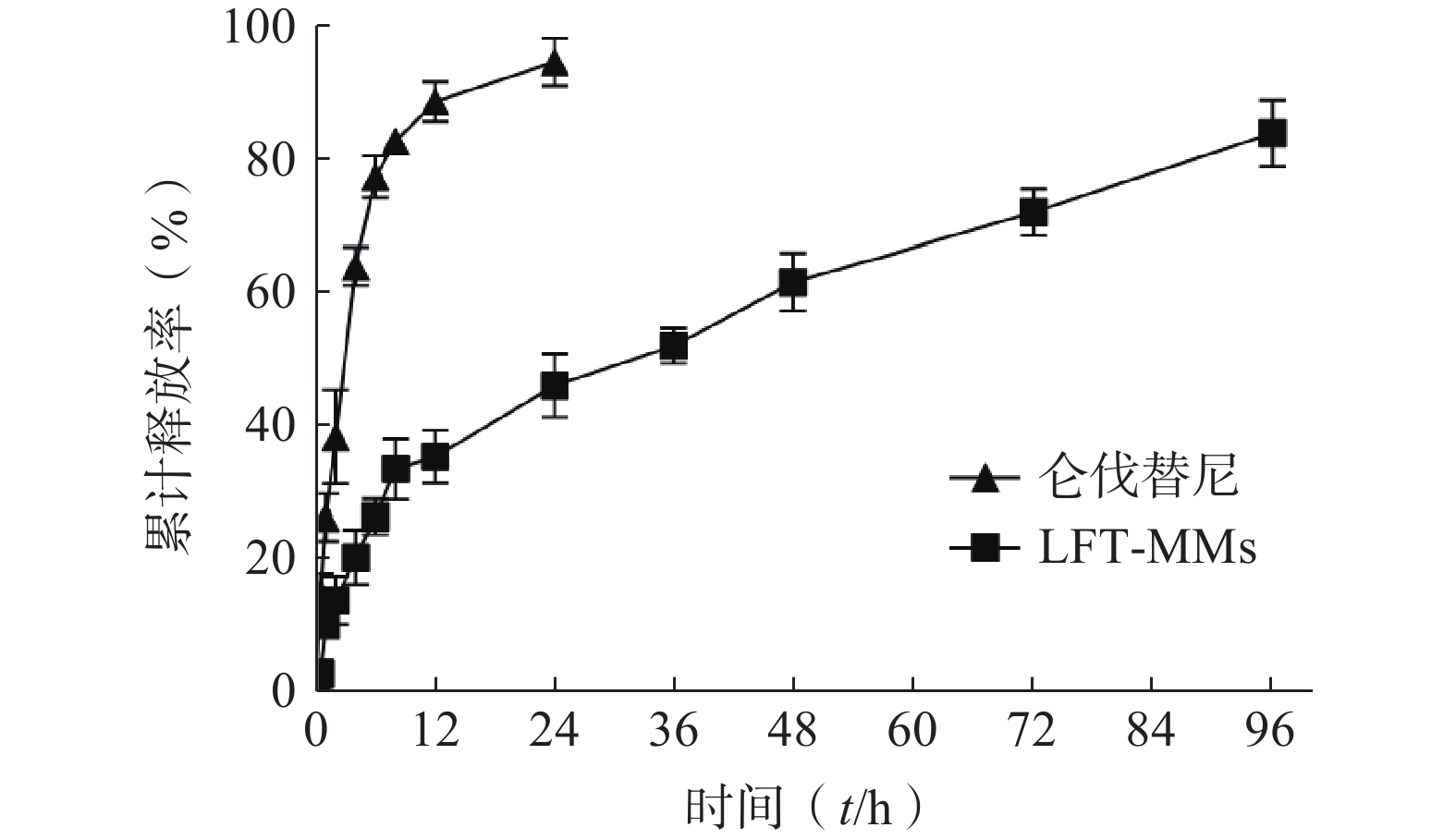
选择含0.5%(w/v)Tween 80的PBS(pH=7.4)作为体外释放介质,采用恒温振荡摇床法进行 LFT-MMs纳米胶束的体外释放实验。将游离仑伐替尼、LFT-MMs纳米胶束冻干粉末用灭菌注射用水稀释至1 mg/ml,精密吸取 3 ml装入预先处理过的透析袋(截留分子量为 8 000),两端扎紧后置于已装40 ml 释放介质的50 ml离心管中,将其放入预设好的恒温振荡器中,温度为 37℃,震荡频率为100 r/min。分别于 0.5、1、2、4、6、8、12、24、36、48、72及96 h 取样3 ml,并补入相同温度,相同体积的释放介质,平行3批样品。取出的样品用0.22 μm微孔滤膜过滤,采用HPLC测定释放介质中仑伐替尼含量,计算累积释放百分率,绘制体外累积释放曲线(图8)。结果显示,在96 h 内LFT-MMs 体外释放缓慢平稳,具有一定的缓释能力。
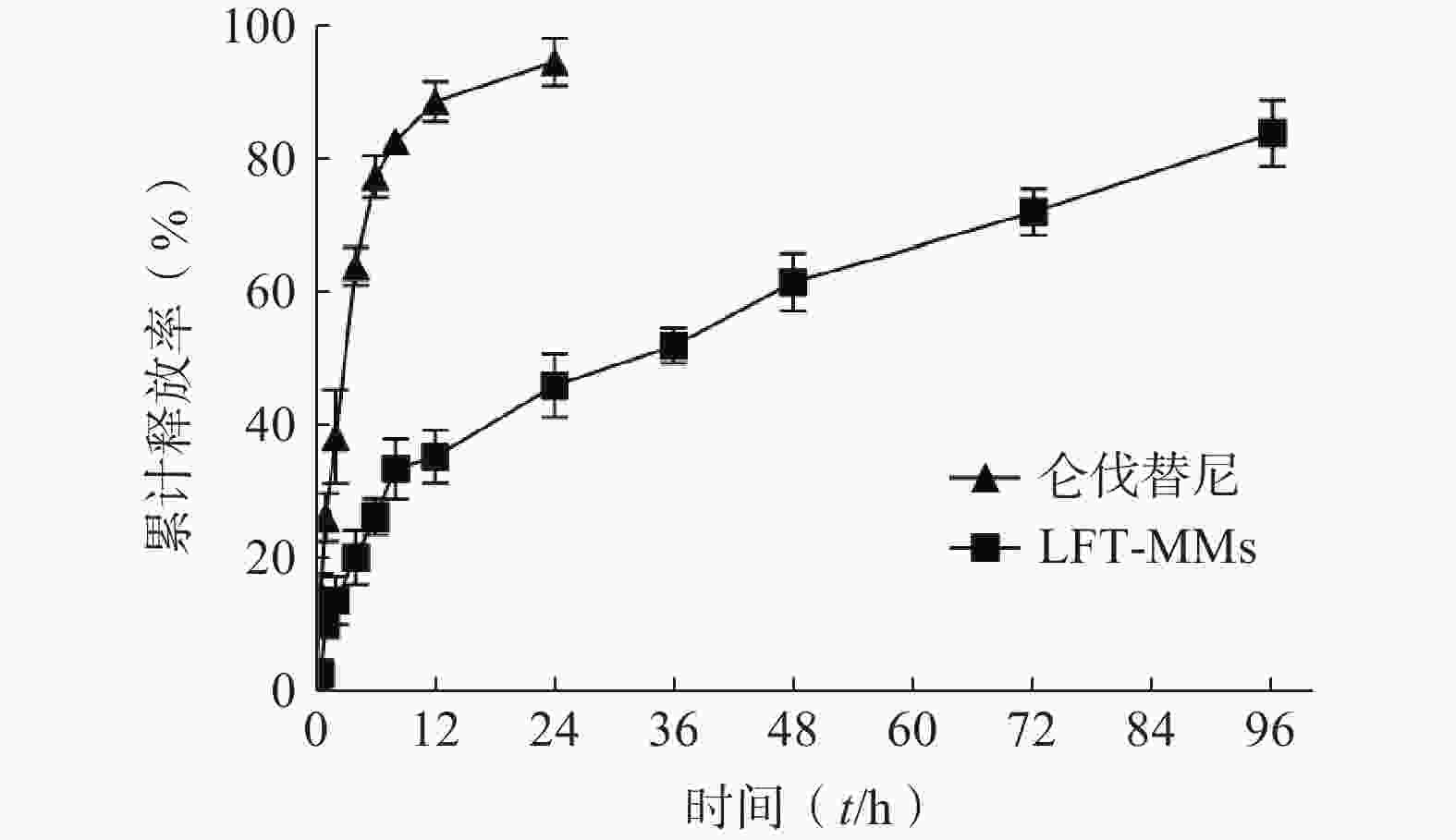
图 8 LFT-MMs 的体外释放曲线(n=3,mean±SD)
-
尽管用于治疗肝脏疾病的化疗药物在不断研发与应用,但由于其水溶性低,对其他正常组织产生毒副作用等问题限制了其临床应用。因此,提升难溶性药物的溶解度和生物利用度在药物制剂设计中至关重要。胶束不仅可以提升药物的溶解度和生物利用度,还可以通过调节其组成和结构来优化药物释放特性,因而在药物递送、基因传递和诊断成像等众多生物医学领域中发挥了重要作用。本研究采用薄膜水化法制备的载仑伐替尼的纳米胶束制剂,具有均一的粒径、良好的分散性和高稳定性。在本研究中,通过单因素结合星点设计-效应面法筛选包括P123质量百分比、载体材料用量、投药量、水化体积、水化时间、旋蒸温度等变量,以EE和DL为考察指标,最终确定了最优处方。在此条件下的验证试验显示,实际EE和DL与预测值接近,证明了利用星点设计-效应面法优化后的胶束制备工艺的稳定性和良好的工艺重现性。
综上所述,本研究通过采用星点设计-效应面法优化了仑伐替尼混合胶束的处方和制备工艺。该方法预测性强,成功制备的胶束具有较高的DL和EE,外观规整,粒径分布均一,且药物释放稳定,为仑伐替尼新剂型的进一步研发提供了重要参考。随着各种新型两亲性嵌段共聚物的不断发展,基于胶束的药物递送系统在从理论到临床应用的转化上将展现出更大的潜力。
Optimization of the preparation process for lenvatinib mixed micelles by central composite design-response surface methodology
-
摘要:
目的 优化仑伐替尼混合胶束的处方及其制备工艺。 方法 以Pluronic P123和F127作为载体材料,通过薄膜水化法制备仑伐替尼混合胶束。利用单因素实验和星点设计-效应面法筛选出最优处方并对其物理性质进行初步表征。 结果 优化后的最佳处方和工艺条件为:P123质量百分比80%、载体材料用量90 mg、投药量10 mg、水化体积6 ml、水化时间45 min、旋蒸温度55℃。制备得到的仑伐替尼混合胶束的平均粒径为(104.0±0.32) nm,PDI为0.22±1.19,Zeta电位为(−2.56±0.81) mV,平均包封率为83.33%±0.30%,平均载药量为8.67%±0.07%。胶束形态为分布均一的规整球形,并显示出一定的缓释性能。 结论 该研究开发的制备工艺简单可行,所得载药胶束具有较高的载药量和包封率且释放稳定,为仑伐替尼混合胶束的进一步研究和开发提供了有价值的参考。 Abstract:Objective To optimize the formulation and preparation process of lenvatinib mixed micelles. Methods Hybrid micelles of lenvatinib were prepared by film hydration method, with Pluronic P123 and F127 as carrier materials. Optimal formulation was selected through single-factor experiments and central composite design-response surface methodology, and preliminary characterization of its physical properties was conducted. Results The optimized formulation and process conditions were identified as follows: P123 mass percentage of 80%, carrier material amount of 90 mg, drug loading of 10 mg, hydration volume of 6 ml, hydration time of 45 min, and rotary evaporation temperature of 55℃. The resulting lenvatinib mixed micelles had an average particle size of (104.0±0.32) nm, a polydispersity index (PDI) of 0.22±1.19, and a Zeta potential of (−2.56±0.81) mV. The average encapsulation efficiency was 83.33%±0.30% and the average drug loading was 8.67%±0.07%. The micelles displayed a uniform spherical morphology with a certain sustained-release capability. Conclusion The preparation process developed in this study was simple and feasible and produced drug-loaded micelles with high drug loading and encapsulation rates, and stable release, which could provide valuable insights for further research and development of lenvatinib mixed micelles. -
图 3 因素对载药量的等高线和效应面图
A.P123 质量百分比(a)、P123-F127 混合载体用量(b) 对载药量的等高线图;B.P123 质量百分比(a)、水相体积(c)对载药量的等高线图;C.P123-F127 混合载体用量(b)、水相体积(C)对载药量的等高线图;D.P123 质量百分比(a)、P123-F127 混合载体用量(b) 对载药量的效应面图;E.P123 质量百分比(a)、水相体积(c)对载药量的的效应面图;F.P123-F127 混合载体用量(b)、水相体积(c)对载药量的效应面图
图 4 因素对包封率的等高线和效应面图
A.P123 质量百分比(a)、P123-F127 混合载体用量(b) 对包封率的等高线图;B.P123 质量百分比(a)、水相体积(c)对包封率的等高线图;C.P123-F127 混合载体用量(b)、水相体积(c)对包封率的等高线图;D.P123 质量百分比(a)、P123-F127 混合载体用量(b) 对包封率的效应面图;E.P123 质量百分比(a)、水相体积(c)对包封率的的效应面图;F.P123-F127 混合载体用量(b)、水相体积(c)对包封率的效应面图
表 1 星点设计因素水平表
因素 P123质量百分比
(%)F127-P123用量
(m/mg)水化体积
(V/ml)−1.68 6.25 49.09 2.93 −1.00 25.00 90.00 6.00 0 52.50 150.00 10.50 1.00 80.00 210.00 15.00 1.68 98.75 250.91 18.07  下载: 导出CSV
下载: 导出CSV
表 2 星点设计表及效应值
序号 a:P123质量
百分比(%)b:载体材料
用量(m/mg)c:水化体积
(V/ml)包封率
(%)载药量
(%)1 80.00 90.00 6.00 90.79 9.53 2 52.50 150.00 10.50 80.95 5.95 3 52.50 150.00 10.50 77.21 6.19 4 25.00 90.00 6.00 59.43 4.91 5 52.50 150.00 10.50 79.97 6.16 6 98.75 150.00 10.50 90.04 5.62 7 80.00 210.00 6.00 77.86 6.96 8 25.00 90.00 15.00 43.77 4.98 9 80.00 210.00 15.00 94.30 4.10 10 52.50 150.00 10.50 84.36 6.82 11 6.25 150.00 10.50 79.19 6.46 12 25.00 210.00 6.00 85.31 3.62 13 52.50 150.00 18.07 76.50 4.33 14 52.50 150.00 10.50 83.22 6.96 15 52.50 250.90 10.50 80.85 3.33 16 52.50 150.00 10.50 82.82 6.99 17 80.00 90.00 15.00 68.94 7.95 18 25.00 210.00 15.00 83.54 5.29 19 52.50 49.09 10.50 52.12 8.99 20 52.50 150.00 2.93 70.97 8.09
下载: 导出CSV
表 3 效应面拟合模型对包封率和载药量的方差分析
方差来源 自由度 包封率 载药量 平方和 均方 F值 P值 平方和 均方 F值 P值 模型 9 2833.07 314.79 12.95 <0.01 41.61 4.62 3.48 0.03 a 1 446.49 446.49 18.37 <0.01 5.08 5.08 3.82 0.08 b 1 1169.85 1169.85 48.12 <0.01 20.96 20.96 15.78 0.00 c 1 13.42 13.42 0.55 0.47 5.96 5.96 4.49 0.06 ab 1 354.05 354.05 14.56 <0.01 3.70 3.70 2.79 0.13 ac 1 18.06 18.06 0.74 0.41 4.77 4.77 3.59 0.09 bc 1 340.34 340.34 14.00 <0.01 0.01 0.01 0.01 0.92 a² 1 25.39 25.39 1.04 0.33 0.62 0.62 0.47 0.01 b² 1 372.27 372.27 15.31 <0.01 0.40 0.40 0.30 0.01 c² 1 91.46 91.46 3.76 0.08 0.32 0.32 0.24 0.64 残差 10 243.11 24.31 13.28 1.33 失拟项 5 209.21 41.84 6.17 0.03 12.21 2.44 11.44 0.01 净误差 5 33.89 6.78 1.07 0.21 总离差 19 3076.18 54.89
下载: 导出CSV
-
[1] ZHAO Y, ZHANG Y N, WANG K T, et al. Lenvatinib for hepatocellular carcinoma: From preclinical mechanisms to anti-cancer therapy[J]. Biochim Biophys Acta Rev Cancer, 2020, 1874(1):188391. doi: 10.1016/j.bbcan.2020.188391 [2] CHEN C Y, WU S M, LIN Y H, et al. Induction of nuclear protein-1 by thyroid hormone enhances platelet-derived growth factor A mediated angiogenesis in liver cancer[J]. Theranostics, 2019, 9(8):2361-2379. doi: 10.7150/thno.29628 [3] WANG Y J, LIU D F, ZHANG T Y, et al. FGF/FGFR signaling in hepatocellular carcinoma: from carcinogenesis to recent therapeutic intervention[J]. Cancers, 2021, 13(6):1360. doi: 10.3390/cancers13061360 [4] AL-SALAMA Z T, SYED Y Y, SCOTT L J. Lenvatinib: a review in hepatocellular carcinoma[J]. Drugs, 2019, 79(6):665-674. doi: 10.1007/s40265-019-01116-x [5] HATANAKA T, NAGANUMA A, KAKIZAKI S. Lenvatinib for hepatocellular carcinoma: a literature review[J]. Pharmaceuticals, 2021, 14(1):36. [6] FACCIORUSSO A, TARTAGLIA N, VILLANI R, et al. Lenvatinib versus sorafenib as first-line therapy of advanced hepatocellular carcinoma: a systematic review and meta-analysis[J]. Am J Transl Res, 2021, 13(4):2379-2387. [7] VENTURA C, JUNCO M, SANTIAGO VALTIERRA F X, et al. Synergism of small molecules targeting VDAC with sorafenib, regorafenib or lenvatinib on hepatocarcinoma cell proliferation and survival[J]. Eur J Pharmacol, 2023, 957:176034. doi: 10.1016/j.ejphar.2023.176034 [8] CHEN Y Y, WANG C C, LIU Y W, et al. Clinical impact of lenvatinib in patients with unresectable hepatocellular carcinoma who received sorafenib[J]. PeerJ, 2020, 8:e10382. doi: 10.7717/peerj.10382 [9] SUESHIGE Y, SHIRAIWA K, HONDA K, et al. A broad range high-throughput assay for lenvatinib using ultra-high performance liquid chromatography coupled to tandem mass spectrometry with clinical application in patients with hepatocellular carcinoma[J]. Ther Drug Monit, 2021, 43(5):664-671. doi: 10.1097/FTD.0000000000000872 [10] SPAHN S, KLEINHENZ F, SHEVCHENKO E, et al. The molecular interaction pattern of lenvatinib enables inhibition of wild-type or kinase-mutated FGFR2-driven cholangiocarcinoma[J]. Nat Commun, 2024, 15(1):1287. doi: 10.1038/s41467-024-45247-6 [11] TAO M, HAN J, SHI J Y, et al. Application and resistance mechanisms of lenvatinib in patients with advanced hepatocellular carcinoma[J]. J Hepatocell Carcinoma, 2023, 10:1069-1083. doi: 10.2147/JHC.S411806 [12] CHEN S Z, HAO X H, LIANG X J, et al. Inorganic nanomaterials as carriers for drug delivery[J]. J Biomed Nanotechnol, 2016, 12(1):1-27. doi: 10.1166/jbn.2016.2122 [13] JACOB S, NAIR A B, BODDU S H S, et al. The emerging role of lipid nanosystems and nanomicelles in liver diseases[J]. Eur Rev Med Pharmacol Sci, 2023, 27(18):8651-8680. [14] KIM S, OH S M, KIM S Y, et al. Role of adsorbed polymers on nanoparticle dispersion in drying polymer nanocomposite films[J]. Polymers, 2021, 13(17):2960. doi: 10.3390/polym13172960 [15] SAKAI-KATO K, YOSHIDA K, TAKECHI-HARAYA Y, et al. Physicochemical characterization of liposomes that mimic the lipid composition of exosomes for effective intracellular trafficking[J]. Langmuir, 2020, 36(42):12735-12744. doi: 10.1021/acs.langmuir.0c02491 [16] SINANI G, DURGUN M E, CEVHER E, et al. Polymeric-micelle-based delivery systems for nucleic acids[J]. Pharmaceutics, 2023, 15(8):2021. doi: 10.3390/pharmaceutics15082021 [17] MOHAN A, NAIR S V, LAKSHMANAN V K. Polymeric nanomicelles for cancer theragnostics[J]. Int J Polym Mater Polym Biomater, 2018, 67(2):119-130. doi: 10.1080/00914037.2017.1309540 [18] JIN G W, REJINOLD N S, CHOY J H. Multifunctional polymeric micelles for cancer therapy[J]. Polymers, 2022, 14(22):4839. doi: 10.3390/polym14224839 [19] MANJAPPA A S, KUMBHAR P S, PATIL A B, et al. Polymeric mixed micelles: improving the anticancer efficacy of single-copolymer micelles[J]. Crit Rev Ther Drug Carrier Syst, 2019, 36(1):1-58. doi: 10.1615/CritRevTherDrugCarrierSyst.2018020481 [20] PATHAN T, GIRASE M, RAY D, et al. Scrutinizing micellar transitions and interfacial properties in mixed micelles comprising sodium dodecyl sulfate and sodium oleate: a tensiometric and scattering insight[J]. J Mol Liq, 2024, 397:124138. doi: 10.1016/j.molliq.2024.124138 [21] LI C L, GUAN H, LI Z H, et al. Study on different particle sizes of DOX-loaded mixed micelles for cancer therapy[J]. Colloids Surf B Biointerfaces, 2020, 196:111303. doi: 10.1016/j.colsurfb.2020.111303 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 8446
- HTML全文浏览量: 4474
- PDF下载量: 27
- 被引次数: 0
