

-
腹主动脉瘤(AAA)是一种严重的慢性炎症性退行性主动脉疾病,特征是腹主动脉壁进行性病理性扩张[1-2]。当局部扩张的腹主动脉直径>正常值的50%时(人体中,最大直径≥30 mm),即可诊断为AAA[3]。大多数AAA患者没有症状,通常缓慢发展,然而也可能迅速进展为大AAA并破裂,死亡率极高[4]。研究表明,每年全球约20万人死于腹主动脉瘤体破裂[5]。高龄、男性、高血压、吸烟、动脉粥样硬化、家族史和环境污染均会增加AAA的患病风险[6-9]。目前大AAA(人体中,最大直径>50 mm)的唯一治疗方法就是开放性或血管内动脉修补术,而对于小AAA(人体中,最大直径<50 mm),尚无明确可以延缓其进展的药物[5]。因此进一步明确AAA的分子调控机制,寻找潜在的药理学干预靶点至关重要。
α7烟碱乙酰胆碱受体(α7nAChR)是一种由5个相同的α7亚单位组成的配体门控离子通道型受体[10]。作为“胆碱能抗炎通路(CAP)”的核心,参与了机体的急慢性炎症过程[11-12]。研究表明,激活α7nAChR在多种心脑血管疾病(如心肌缺血再灌注、缺血性脑卒中)中发挥保护作用[13-14]。主动脉细胞外基质(ECM)降解、血管平滑肌细胞(VSMC)凋亡和血管壁慢性炎症是AAA发病机制的重要病理过程[15-17]。炎症细胞释放的细胞因子不仅会加剧ECM降解,还会导致VSMC细胞凋亡。多种抗炎药物如塞来昔布、c-Jun N-末端激酶抑制剂和缓激肽2受体拮抗剂等均已被证明可以减缓小鼠AAA的进展并降低其破裂的风险[18]。因此抑制炎症反应已被推测是治疗AAA的可行手段[1]。
由此,我们推设激活α7nAChR可通过抑制炎症反应减轻CaCl2诱导的小鼠AAA损伤。本研究通过构建CaCl2诱导的小鼠AAA模型及体外细胞实验来研究激活α7nAChR对AAA的作用及可能机制。
-
α7nAChR基因敲除(α7nAChR−/−)和野生型(WT)小鼠由同济大学附属第十人民医院赠送,其饲养与繁育工作于内蒙古医科大学SPF级实验动物室完成。SD大鼠购买自内蒙古医科大学实验动物中心,用于VSMC的提取。实验进行期间,动物房温度维持在22~25℃,湿度保持在70%左右,且保持7:00~19:00的照明状态。所有动物均能够自由饮水、饮食,且笼内能够循环通风。本实验过程中所有动物操作,均符合内蒙古医科大学动物中心的管理规范。
-
TNF-α抗体(BA0131,Boster公司);IL-1β抗体(ab205924)、Gasdermin D(GSDMD)抗体(ab209845)、基质金属蛋白酶2(Matrix Metalloproteinases2,MMP2)MMP2 抗体(ab92536) (abcam公司);NOD-like receptor thermal protein domain associated protein 3(NLRP3)抗体 (#15101)、cleaved Caspase 1抗体(#89332)(cst公司);GAPDH抗体、β-actin抗体、DAPI染色液(碧云天生物技术公司);Donkey anti-mouse荧光二抗、Donkey anti-rabbit 荧光二抗(Li-cor公司);PNU-282987、戊巴比妥钠、氯化钙(Sigma公司);动物组织DNA抽提试剂盒、琼脂糖、α7nAChR敲除鼠鉴定引物、胶原酶(Ⅱ型)(上海生工生物公司);Taq酶(Takara公司);NaRed 染料(北京派拓科技有限公司);组化试剂盒DAB显色剂(DAKO公司);OCT包埋剂(Sakura公司);EVG染液套装(Service biology公司);蛋白Marker、BCA蛋白测定试剂盒(Thermo Scientific公司);伊红染液、苏木精染液(武汉谷歌生物公司);PBS缓冲液、50 X TAE溶液(上海博光生物有限公司);FBS、胰蛋白酶(Gibco公司);高糖DMEM培养基(Hyclone公司);TNF-α重组蛋白(Peprotech公司)。
-
Odssey红外荧光显像系统(Li-Cor公司);全自动酶标仪、二氧化碳细胞培养箱(Thermo Scientific公司);包埋机、切片机(武汉俊杰电子有限公司);超净工作台(苏州净化有限公司);台式离心机、普通PCR仪(Eppendorf公司);琼脂糖凝胶电泳仪(Tanon公司);生物电泳图像分析系统(上海复日科技有限公司)。
-
剪取小鼠鼠尾约4 mm,加入组织裂解液,56℃水浴1~3 h使鼠尾裂解完全。用动物组织DNA抽提试剂盒进行小鼠DNA的提取。将DNA溶液与Taq酶、α7nAChR的3种引物及水配制成扩增体系,于PCR仪上扩增。
-
称取适量琼脂糖,加入1 XTAE溶液后于微波炉加热使其完全溶解,在冷却之前加入NaRed染料,混匀,倒入配胶装置并插入加样孔梳。待完全冷却后拔出梳子,加样后开始电泳。电泳结束后在生物电泳图像分析系统仪器上成像并拍照分析。
-
7~8周龄的雄性WT小鼠及α7nAChR−/−小鼠随机分为4组,分别为野生型对照组(WT)、敲除鼠对照组(α7nAChR−/−)、野生型模型组(WT+CaCl2)和敲除鼠模型组(α7nAChR−/−+CaCl2)。使用戊巴比妥钠(50 mg/kg,i.p.)麻醉小鼠后,开腹,充分暴露腹主动脉。对肾动脉分支以下至髂总动脉段的腹主动脉进行充分游离,用生理盐水(对照组)或0.5 mol/L的CaCl2溶液(模型组)浸泡后的无菌纱布完全包裹腹主动脉外膜15 min,随后将残留的CaCl2溶液使用生理盐水冲洗,关腹,缝合。术后3 d内于腹部伤口处涂抹青霉素以防感染。28 d后麻醉处死小鼠,获取主动脉标本。小鼠主动脉最大直径扩张1.5倍即判定主动脉瘤形成,即造模成功。
-
固定后的主动脉标本,使用梯度酒精脱水后进行石蜡包埋,切片机连续切片,厚度约4 μm/片。将石蜡切片脱蜡处理后再水化,进行HE染色;染色封片后置于显微镜下观察,评估组织学特征。对脱蜡后再水化的切片进行EVG染色以评估主动脉壁的弹性蛋白完整性。
-
将石蜡切片脱蜡处理后与一抗(TNF-α,IL-1β,NLRP3,GSDMD;按照说明书推荐比例配制成相应溶液)于4℃孵育过夜,PBS缓冲液洗涤切片,随后加入相应二抗孵育1 h,再次用PBS缓冲液洗涤后用DAPI对细胞核进行染色。染色结束后将切片置于显微镜下观察并评估抗体表达水平。
-
SD大鼠麻醉后开腹获取主动脉。转移至超净台,用胶原酶溶液于37℃细胞培养箱中消化30 min,取出主动脉用PBS缓冲液清洗后置于含20% FBS的高糖DMEM培养基中,于37℃细胞培养箱中培养24 h。随后用PBS缓冲液清洗后将其置于含有胶原酶和胰蛋白酶的混合消化液中,37℃反应2 h。终止消化后收集组织细胞悬液,离心弃上清液,加入含20% FBS的高糖DMEM培养基重悬细胞,置于37℃继续培养,2~3 d后换液并传代。选用第3~8代细胞用于后续实验。
-
探究TNF-α刺激VSMC不同时间点(0、3、6、12、24、48 h)的炎症因子表达,分为6组,分别为对照组(Con,即TNF-α刺激细胞0 h)、3 h处理组(3 h)、6 h处理组(6 h)、12 h处理组(12 h)、24 h处理组(24 h)和48 h处理组(48 h)。探究PNU-282987激活α7nAChR对VSMC炎症因子表达的影响,分为对照组(Con)、模型组(TNF-α,TNF)和模型加药组(TNF-α+PNU-282987,TNF + PNU)。
-
取各组处理后VSMC细胞,采用BCA法测定蛋白浓度。蛋白经SDS-PAGE分离后转移至PVDF膜,封闭后与一抗NLRP3,MMP2于4℃孵育过夜,洗涤后加入荧光二抗孵育液,室温避光孵育2 h后重复洗涤3次。用Odyssey红外成像系统成像并拍照,结合Quantity One软件分析蛋白表达水平,以β-actin或GAPDH为内参。
-
计量资料以均数±标准误(means±SEM)表示。利用GraphPad Prism 8.0 软件进行统计分析,两组间比较采用非配对T检验,多组之间选用单因素方差分析。P<0.05表示差异有统计学意义。
-
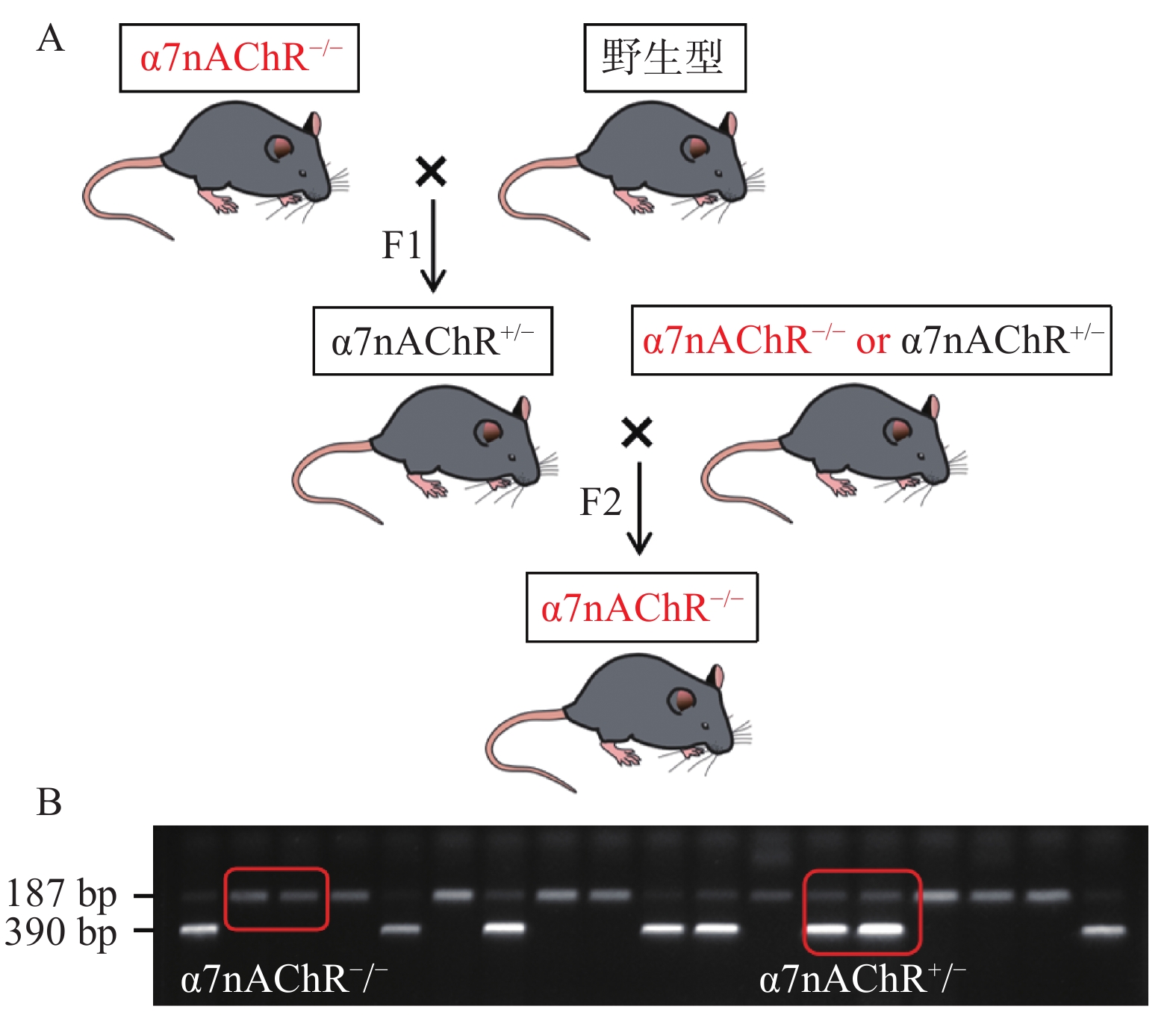
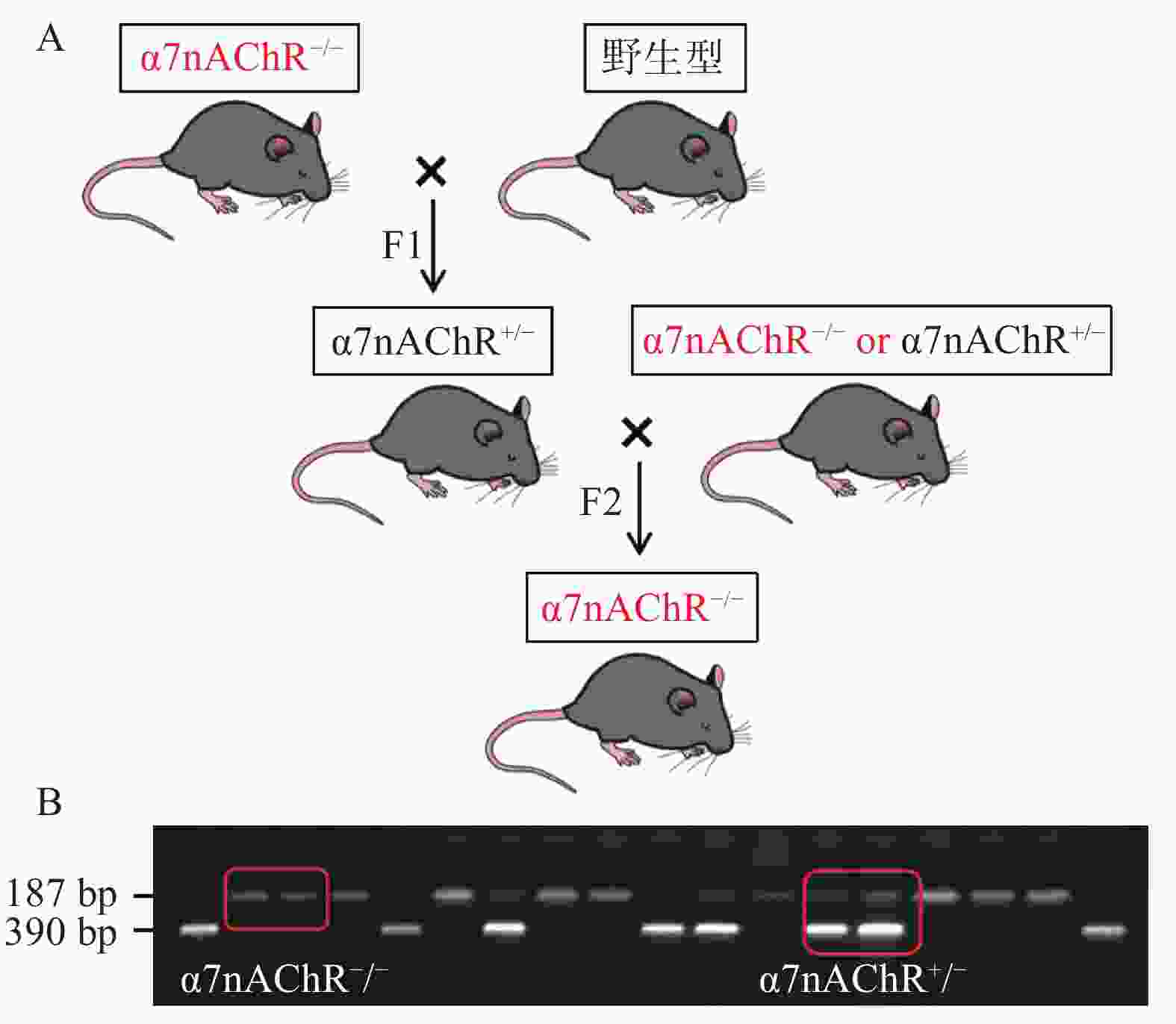
根据以下小鼠繁育模式,我们将α7nAChR−/−小鼠与WT小鼠配种,可得到α7nAChR−/−小鼠、α7nAChR杂合子(α7nAChR+/−)小鼠、WT小鼠;通过获取小鼠尾部DNA并进行琼脂糖凝胶电泳,判定子代小鼠基因型。若在小分子量处出现一条带(187 bp),则为α7nAChR−/−小鼠;若在大分子量处出现一条带(390 bp),则为WT小鼠;若有两个条带(390 bp和187 bp)出现,则为α7nAChR+/−小鼠,见图1。
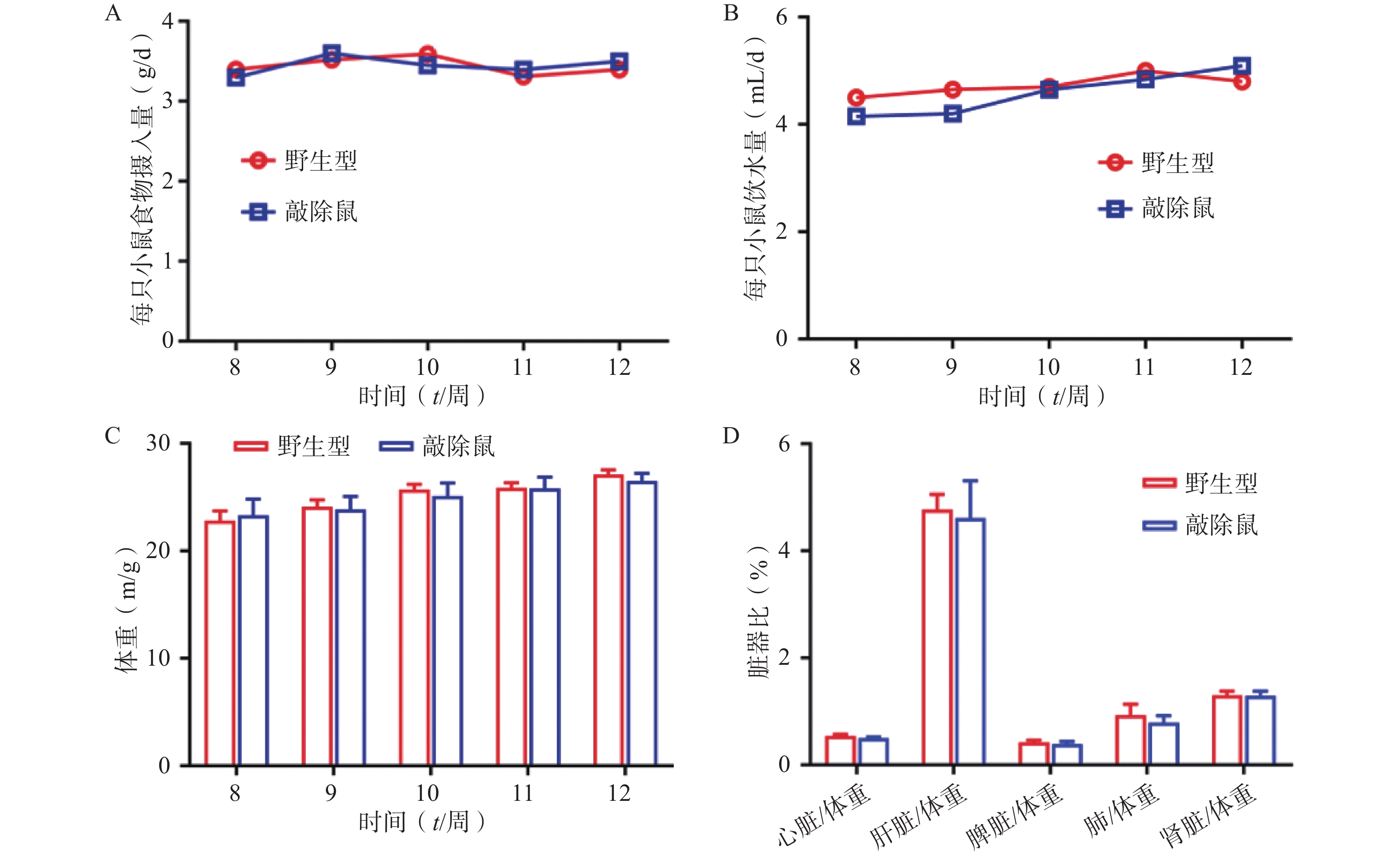
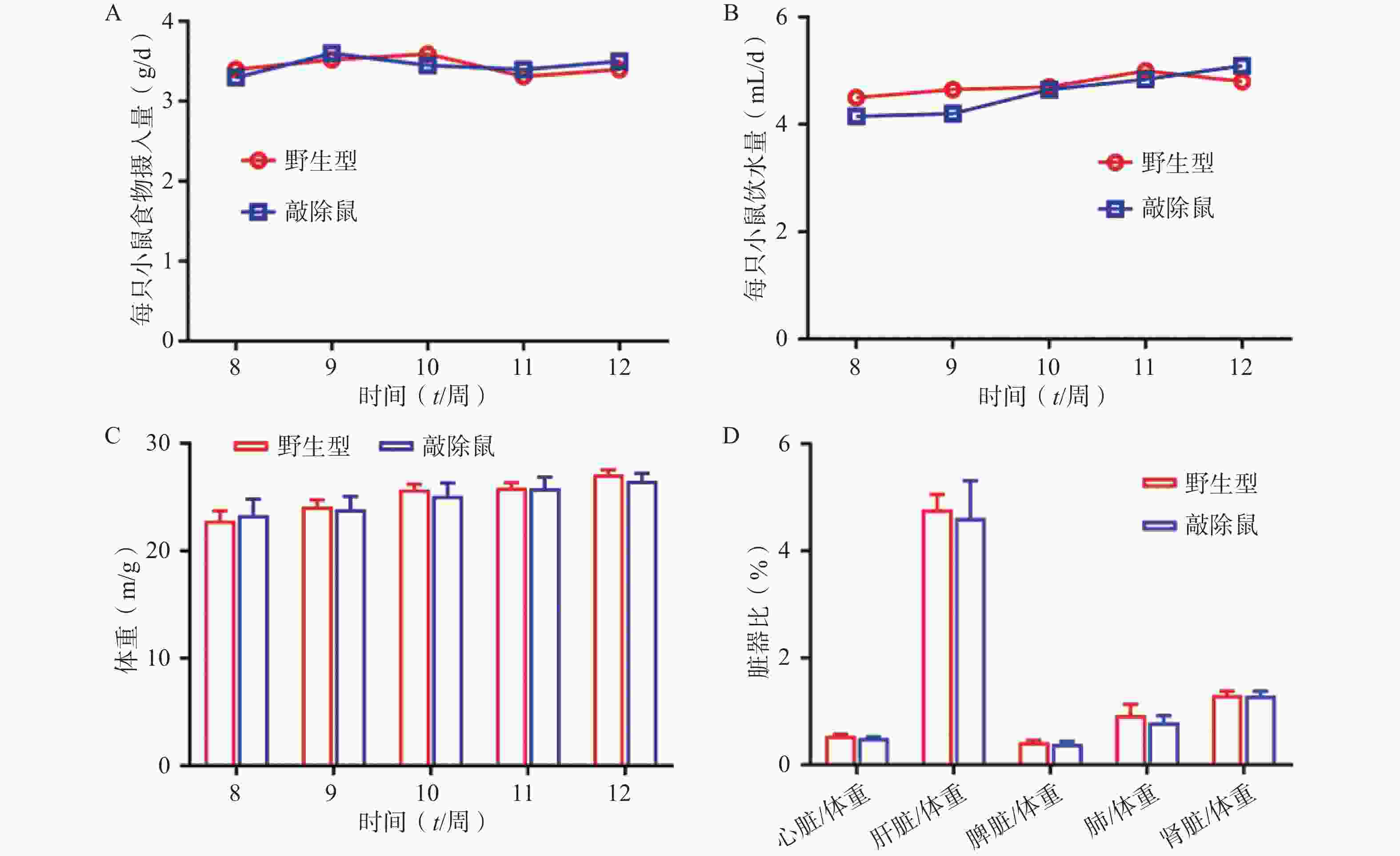
为明确敲除α7nAChR对小鼠基本生理指标是否有影响,我们同步记录α7nAChR−/−小鼠和WT小鼠(8~12 周龄)的进食、饮水量及体重变化;同时选用8周龄α7nAChR−/−小鼠与WT小鼠,检测两者心脏、肝脏、脾脏、肾脏、肺与体重的比值,见图2。结果发现:两组小鼠的体重、进食饮水量及重要脏器体重比均无显著差异。以上结果提示敲除α7nAChR对小鼠基本生理体征无明显影响。
-
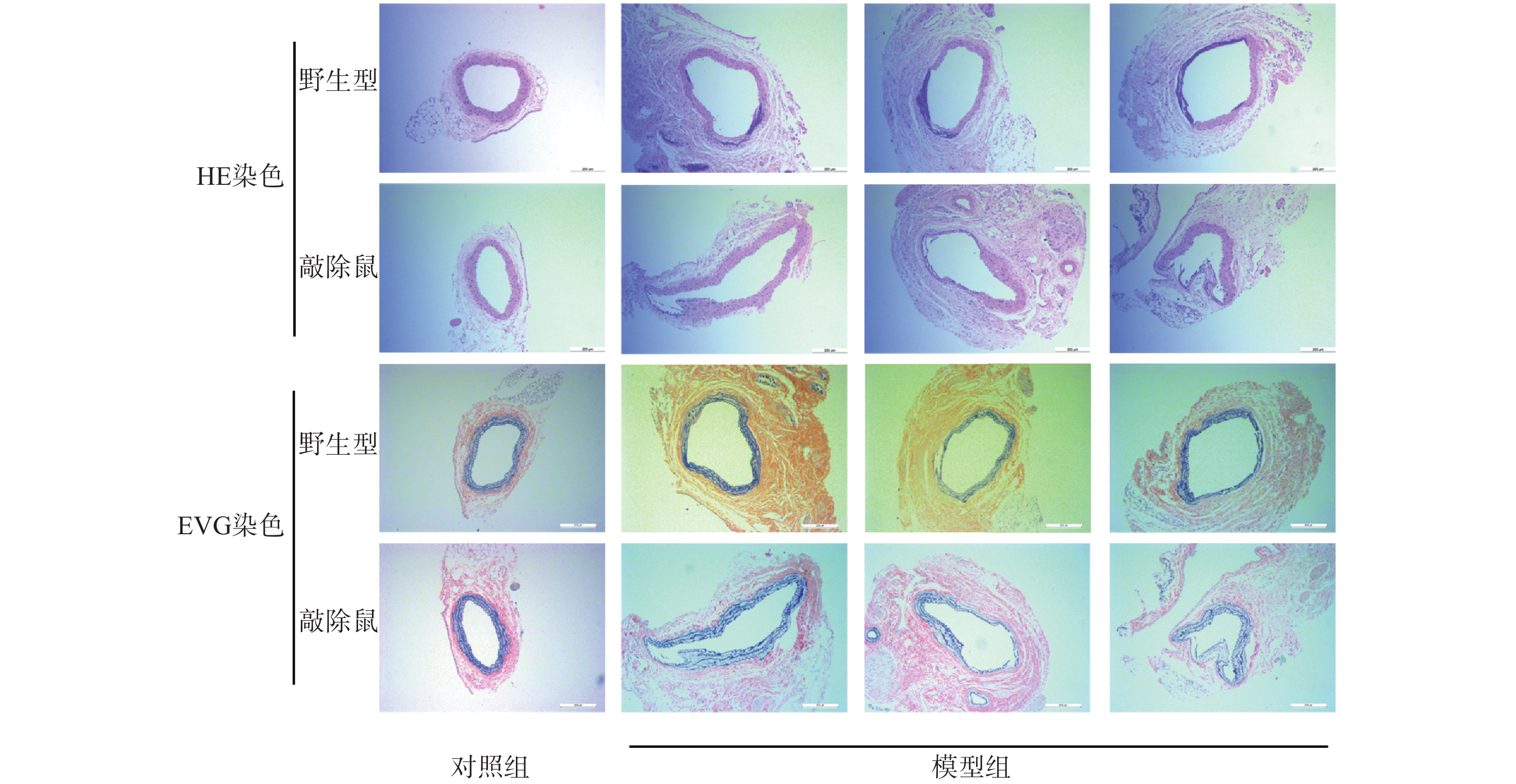
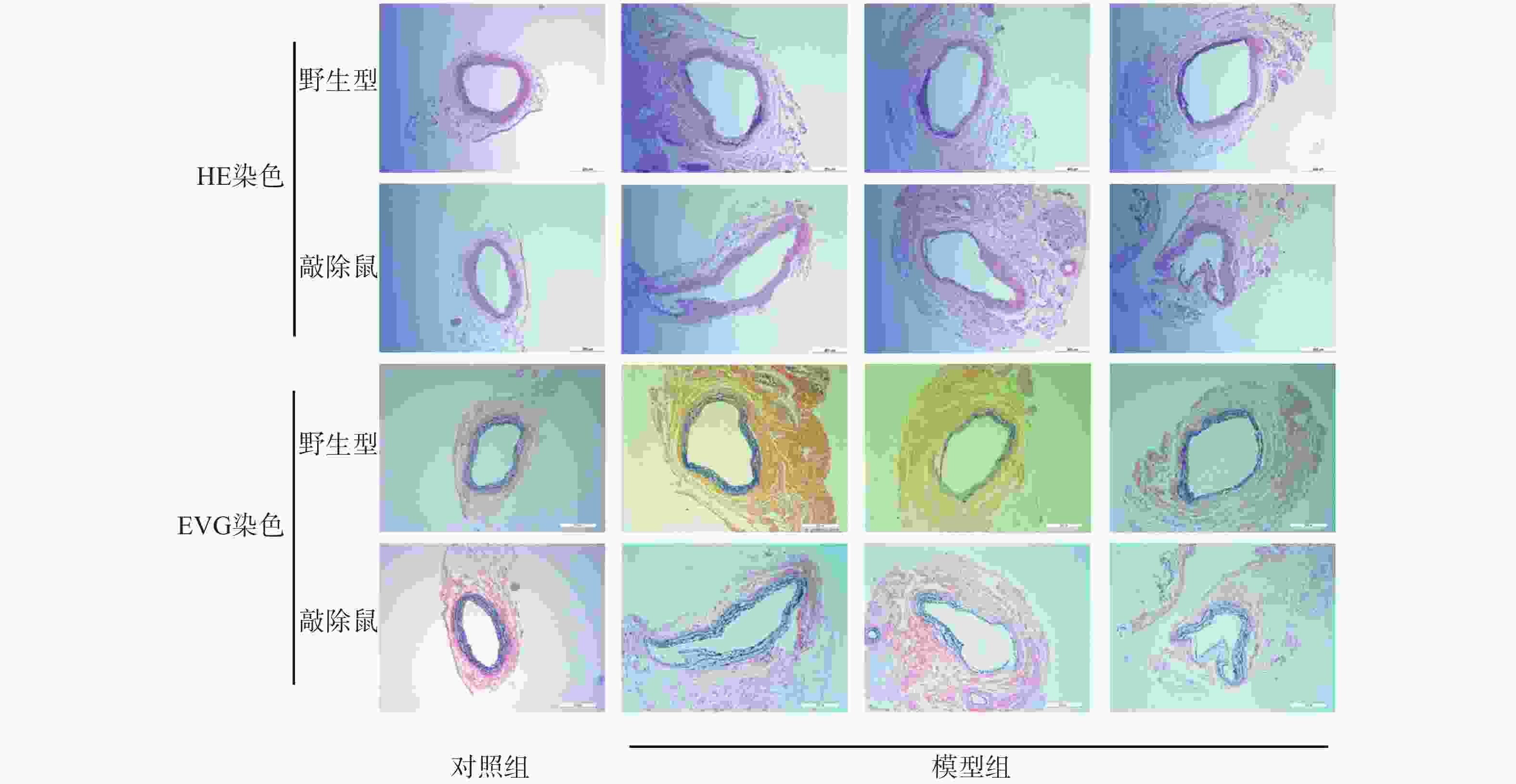
与其他啮齿类动物AAA模型相比,CaCl2诱导的AAA小鼠模型病变部位常位于肾动脉分支下段的腹主动脉,与人类AAA相似[19]。故我们选用CaCl2诱导小鼠形成AAA模型。如图3所示,对照组的WT与KO小鼠主动脉壁的3层组织结构即内膜、中膜和外膜均清晰可见,弹性纤维结构完整,走形规则,厚度均匀。与对照组相比,CaCl2孵育后的AAA小鼠主动脉壁组成结构明显被破坏,中膜的弹性纤维层显著变薄,甚至断裂、消失,中膜部分僵直并失去正常的波浪状结构,有的呈烧焦状。值得注意的是,敲除α7nAChR的小鼠CaCl2孵育后其主动脉的损伤更为严重。
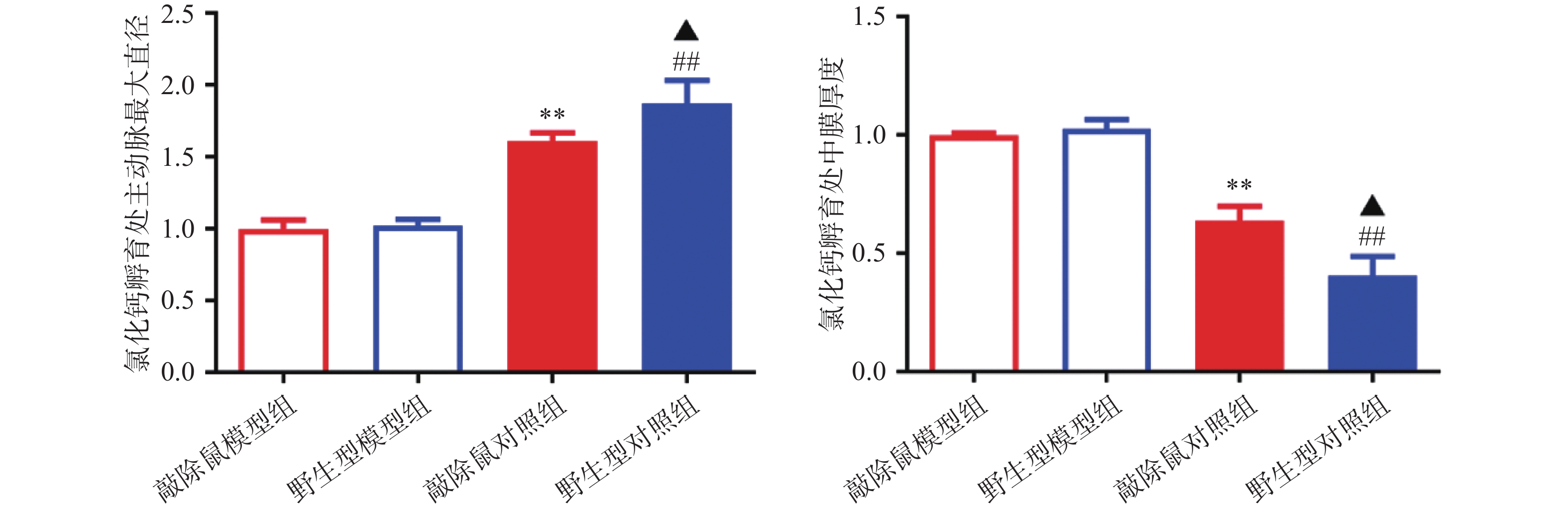
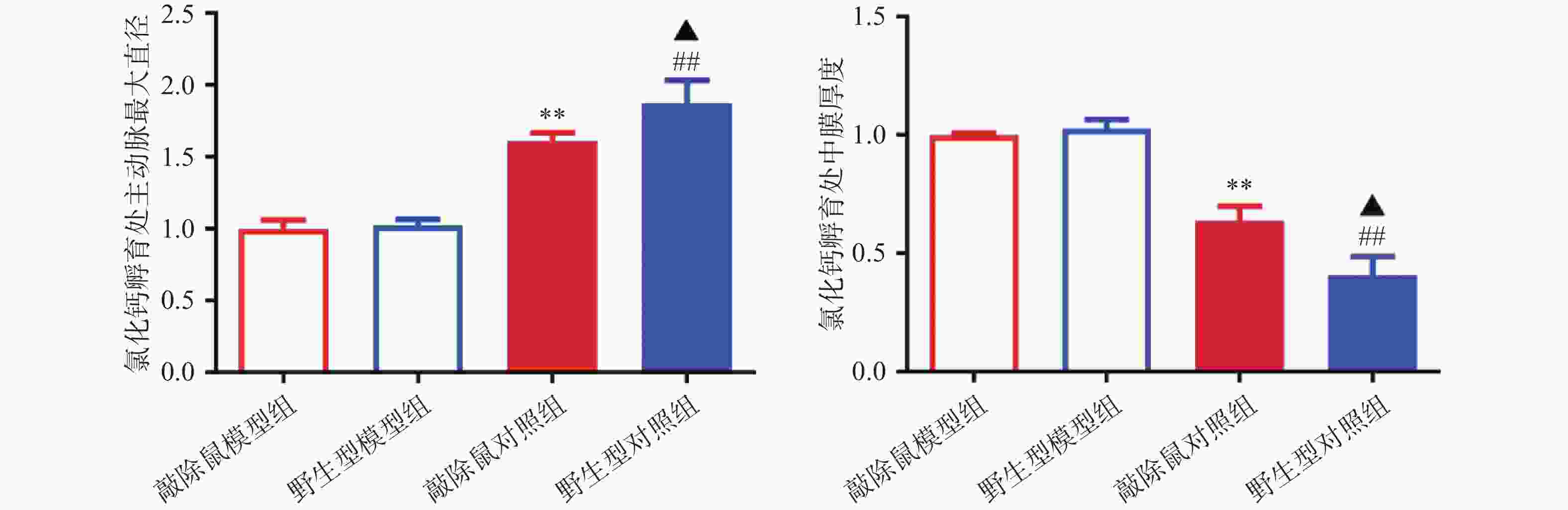
对HE染色结果进行统计分析发现:WT和α7nAChR−/−小鼠的主动脉直径及中膜厚度,无明显差异;CaCl2孵育后,模型组小鼠主动脉明显扩张,中膜明显变薄,证明我们成功构建小鼠AAA模型;与WT+CaCl2小鼠相比,α7nAChR−/−+CaCl2小鼠主动脉进一步扩张,中膜厚度进一步减小,即α7nAChR−/−小鼠AAA模型中腹主动脉损伤更严重,见图4。
-
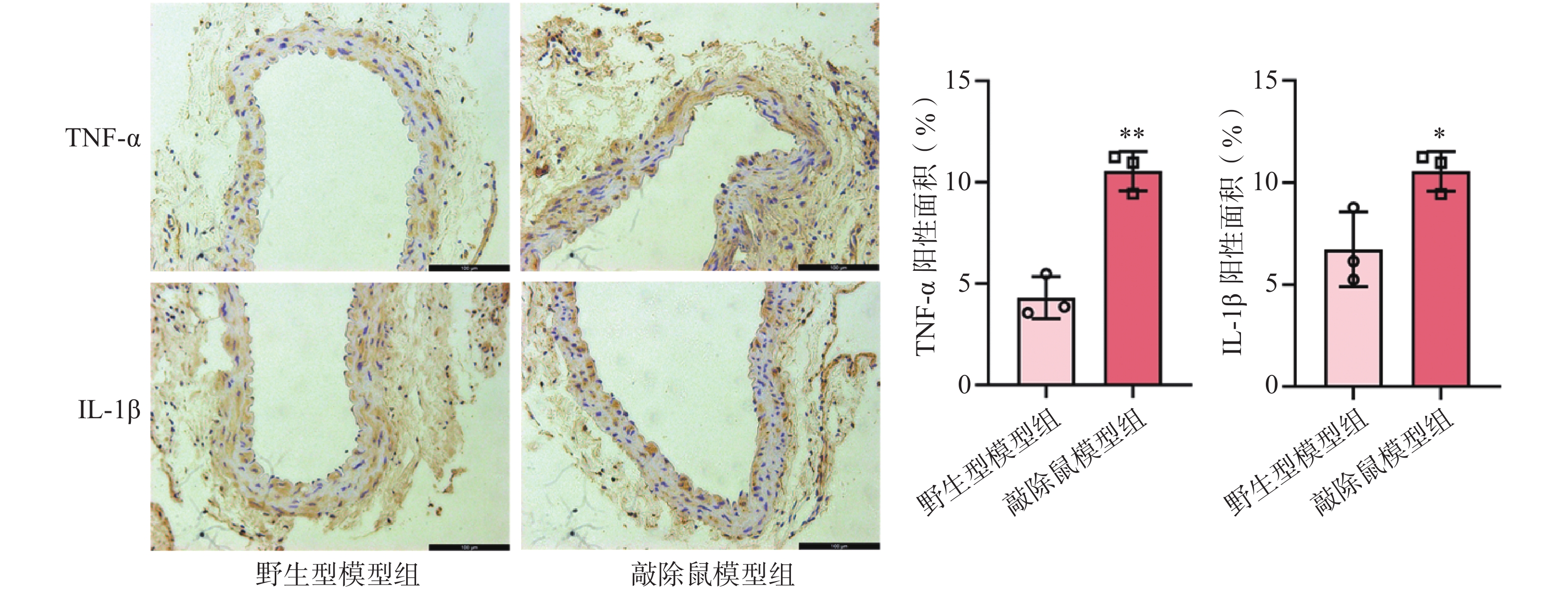
研究认为,血管慢性炎症是腹主动脉瘤形成的核心因素之一[20]。我们前面的研究证明敲除α7nAChR明显加重了AAA小鼠的主动脉损伤。为了验证这一结果是否与加重小鼠血管壁炎症反应相关,我们通过IHC染色检测了各组小鼠主动脉中炎症因子的表达情况。结果显示,CaCl2诱导后,与WT+CaCl2小鼠相比,敲除α7nAChR显著增加了AAA小鼠主动脉中炎症因子TNF-α及IL-1β的表达,见图5。
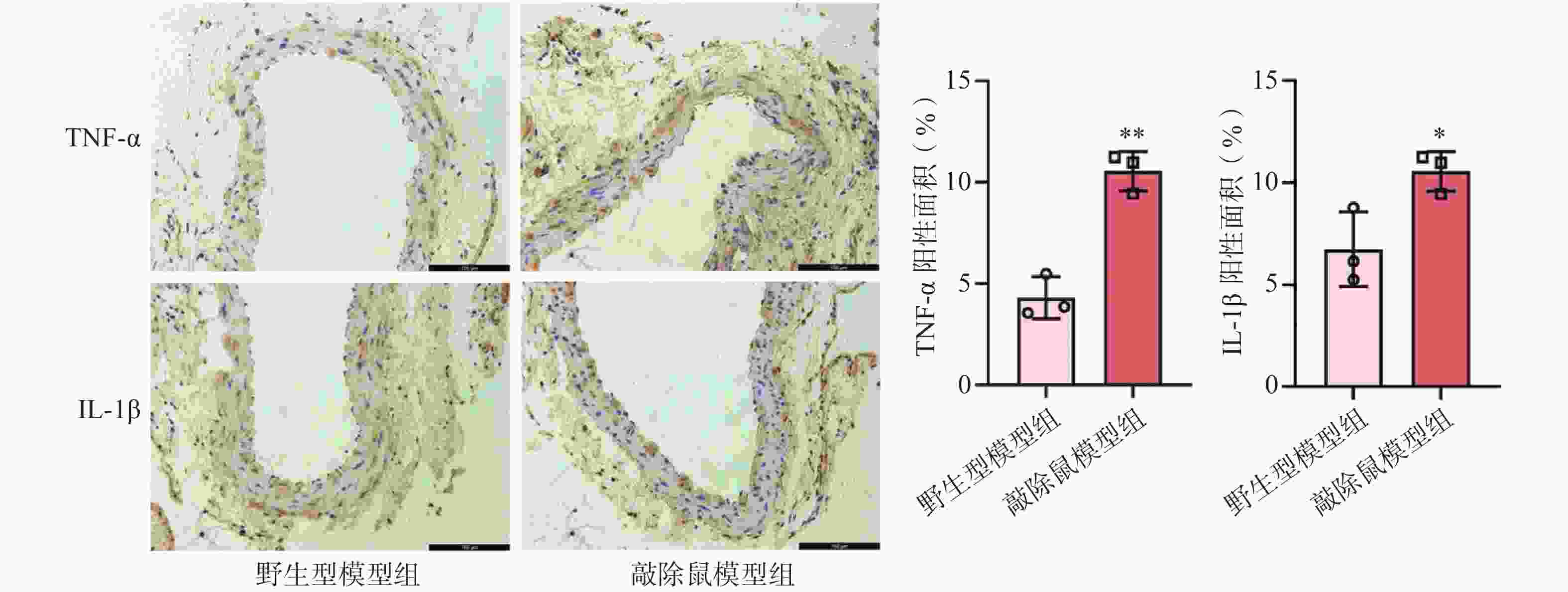
-
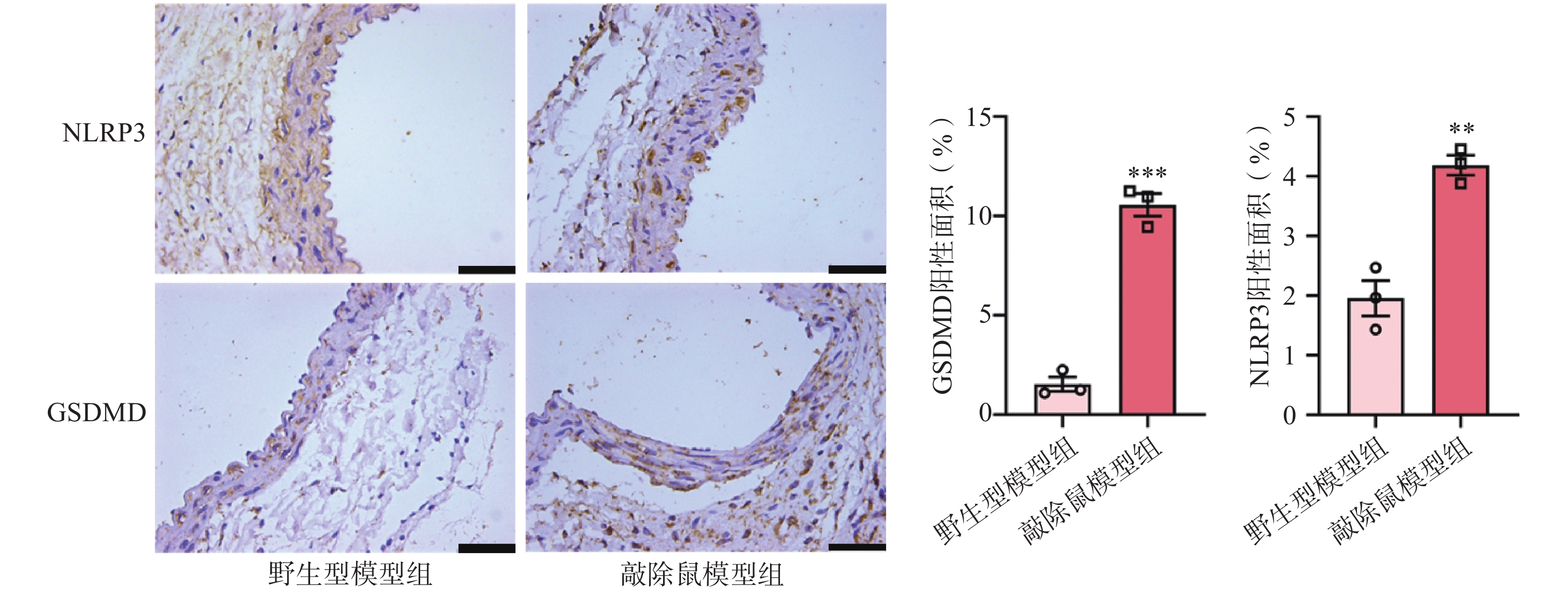
近来,许多研究表明NLRP3炎症小体与AAA的发生发展有一定相关性[21-22]。NLRP3的激活可以促进pro-Caspase-1的自我剪切,GSDMD可以被激活的Caspase-1切割,N-GSDMD通过在细胞膜上形成寡聚孔从而引发细胞焦亡[23]。因此,我们评估了NLRP3和GSDMD在AAA中的表达。IHC染色结果显示,与WT+CaCl2小鼠相比,小鼠缺失α7nAChR后,其主动脉中NLRP3和GSDMD的表达水平明显上调,见图6。这一结果提示敲除α7nAChR使AAA小鼠主动脉损伤更严重,可能是通过激活NLRP3及下游GSDMD进一步促进炎症因子释放所致。
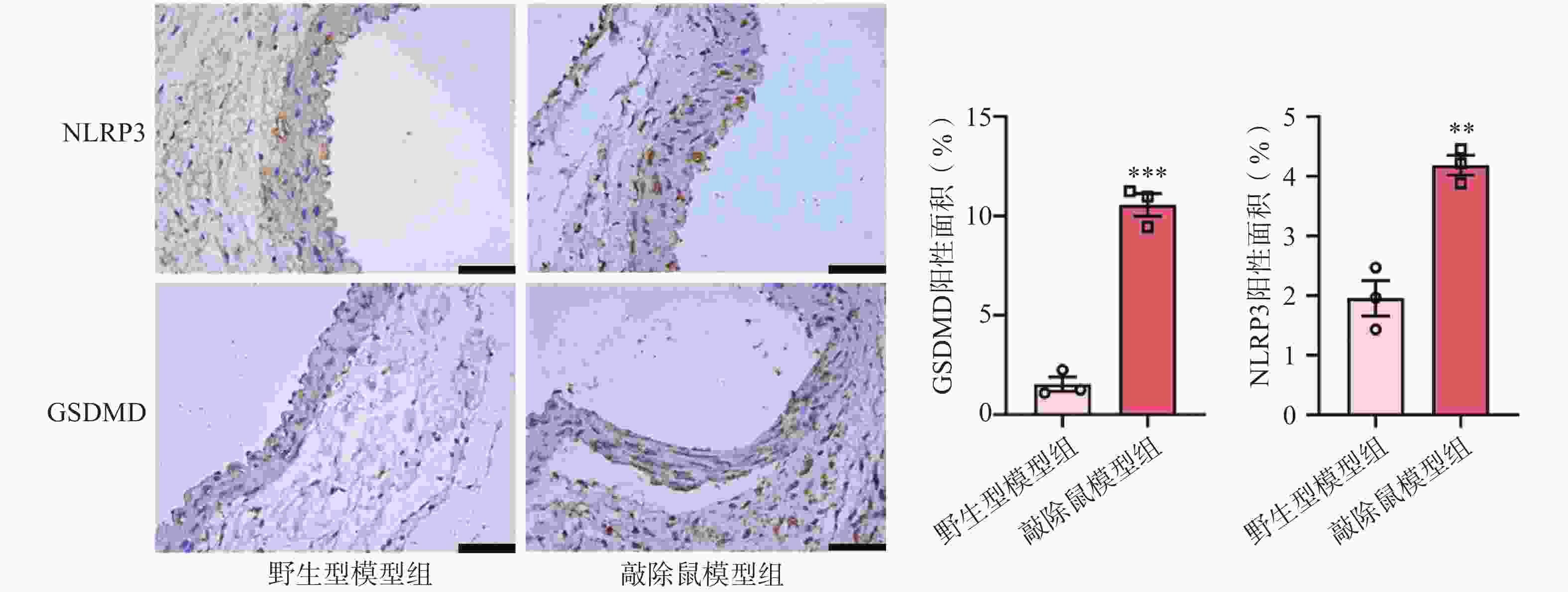
-
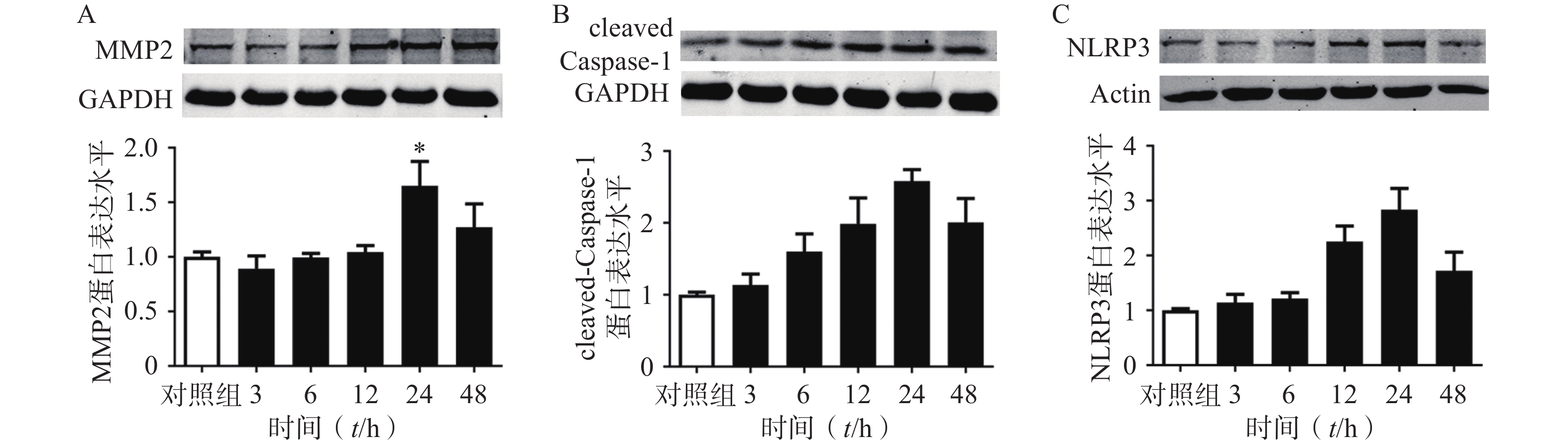
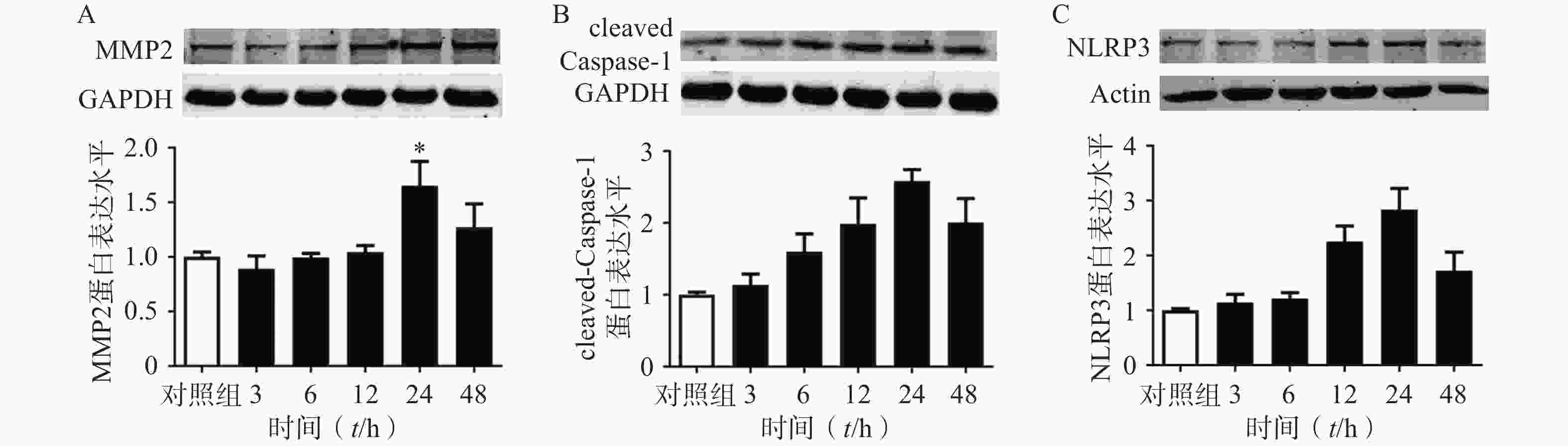
选用100 ng/ml的TNF-α刺激大鼠原代VSMC以模拟AAA血管慢性炎症的微环境[24]。首先我们在大鼠来源的原代VSMC上,明确了TNF-α刺激细胞不同时间对MMP2,cleaved Caspase1,NLRP3蛋白表达的影响。结果表明:随着TNF-α刺激VSMC时间的增加,MMP2,cleaved Caspase1,NLRP3的蛋白表达逐渐增多,在24 h时达到峰值,见图7。因此我们后续实验选择的TNF-α给药时间点为24 h。
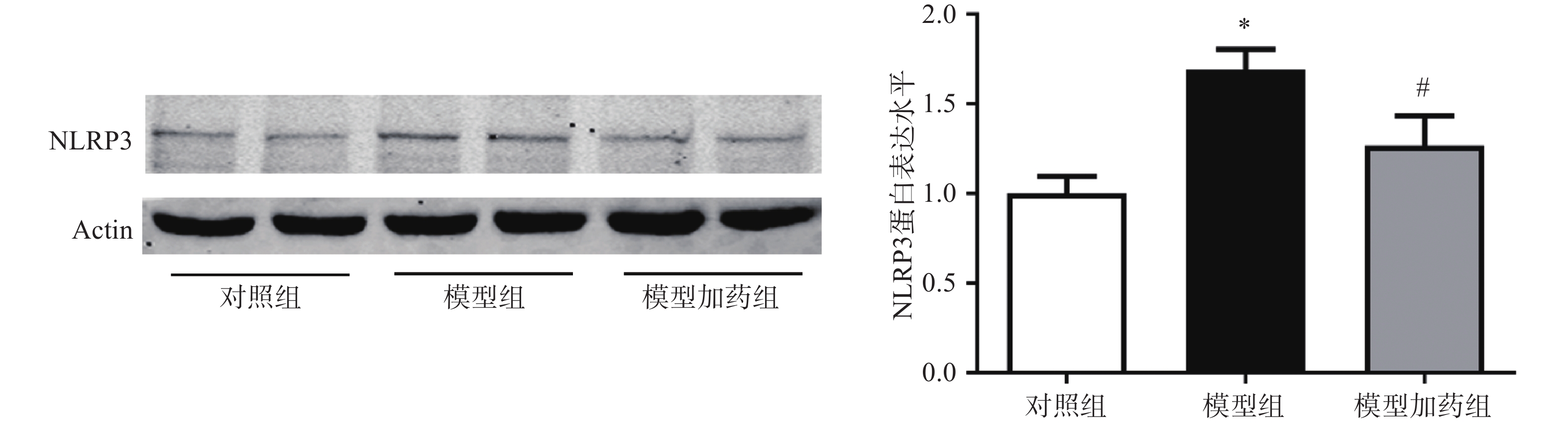
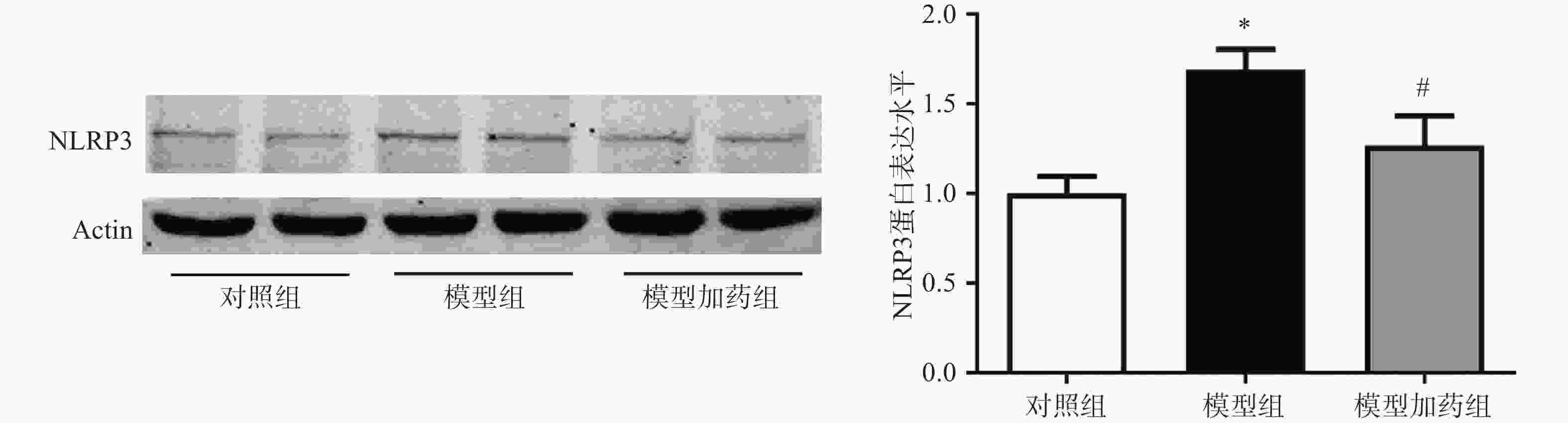
上述体内实验已证明敲除α7nAChR可能通过激活NLRP3炎症小体进一步促进炎症因子的释放而加重AAA小鼠主动脉损伤。因此我们猜测,激活α7nAChR能够抑制NLRP3炎症小体的激活。通过western-Blot检测PNU-282987(10 µmol/L)激活α7nAChR对NLRP3表达的影响。结果显示,与对照组相比,TNF-α模型组NLRP3的表达明显上调,与AAA小鼠中NLRP3表达增加一致;激活α7nAChR能够显著抑制TNF-α诱导的NLRP3表达上调,见图8。
-
AAA一旦破裂,其致死率高达80%以上[25]。近年来,我国逐步迈入老龄化社会,随着人口老龄化的加剧,AAA的患病率也逐年增加[26]。因此探寻减慢AAA扩张、延缓AAA病理进程的药理学干预靶点迫在眉睫。
血管壁慢性炎症在AAA发生发展过程中发挥着十分重要的作用。不论是在动物AAA模型还是人体病变的AAA组织中,均能检测到炎症细胞的浸润,如CD4+T细胞和巨噬细胞[26]。Ang Ⅱ诱导ApoE−/−小鼠形成AAA模型是目前应用最广泛的造模方式,但该模型更接近主动脉夹层的病理特征,且瘤体多发生在肾动脉分支以上部位的主动脉处,与人类不同[19]。而CaCl2诱导的小鼠AAA模型的病变部位常位于肾动脉分支下段的腹主动脉,正是人类AAA的常见部位,且该模型与人类AAA疾病相似的病理特征包括主动脉组织钙化、平滑肌细胞凋亡、蛋白酶活性增强、弹性蛋白被吸收、中膜变薄等[20]。因此,我们选用CaCl2诱导小鼠AAA模型来探究α7nAChR在AAA中的作用。结果表明,CaCl2孵育成功诱导小鼠AAA模型,且与WT小鼠AAA组相比,α7nAChR敲除鼠AAA组的主动脉损伤更为严重,α7nAChR的缺失也进一步加重了AAA小鼠的炎症反应。
最近的研究报道,α7nAChR可通过抑制小胶质细胞的NLRP3炎症小体对小鼠缺血性脑卒中起保护作用[13]。MCC950通过阻断巨噬细胞中NLRP3炎症小体激活而抑制小鼠AAA的形成[27]。众所周知,NLRP3炎症小体的激活可以促进pro-Caspase-1的自我剪切,GSDMD可以被活化的Caspase-1切割,N-GSDMD通过在细胞膜上形成寡聚孔促使炎症因子释放从而引发细胞焦亡[23]。我们猜测NLRP3炎症小体的激活参与了AAA的病理生理进程,并评估了NLRP3及GSDMD在AAA中的表达。研究发现,敲除α7nAChR显著增加了AAA中NLRP3及GSDMD蛋白的表达;激活α7nAChR可抑制TNF-α诱导的VSMC中NLRP3表达的上调。
综上所述,敲除α7nAChR促使炎症因子增多加重CaCl2诱导的小鼠AAA损伤,而激活α7nAChR可减少炎症因子释放减轻小鼠AAA损伤,其机制可能与调控NLRP3/GSDMD通路相关。本实验对激活α7nAChR减轻CaCl2诱导小鼠AAA损伤的可能机制进行了初步探索,相关作用机制仍需更深入的研究。
Amelioration chloride-induced abdominal aortic aneurysm injury by activation of α7nAChR s calcium in mice
doi: 10.12206/j.issn.2097-2024.202409045
- Received Date: 2024-09-20
- Rev Recd Date: 2025-04-16
- Available Online: 2026-07-29
-
Key words:
- α7 nicotinic acetylcholine receptor /
- inflammation /
- CaCl2 /
- abdominal aortic aneurysm
Abstract:
| Citation: | ZHANG Wenjing, FU Hui, GUO Xiaobin, GUO Hao. Amelioration chloride-induced abdominal aortic aneurysm injury by activation of α7nAChR s calcium in mice[J]. Journal of Pharmaceutical Practice and Service. doi: 10.12206/j.issn.2097-2024.202409045

|
 DownLoad:
DownLoad: