

 下载:
下载:
-
血管内皮细胞是沿血管腔表面排列的单层内皮细胞,将血管腔与血管平滑肌及组织分隔开,在维持血管稳态中具有重要的作用。心血管系统中复杂应激环境引起的内皮细胞整合应激反应(ISR)、炎症反应所造成的内皮细胞损伤是动脉粥样硬化的病因[1-2]。与肌组织细胞线粒体相比,内皮细胞线粒体的首要功能是参与信号转导维持细胞功能的完整,其次是进行能量代谢[3-4]。线粒体氧化应激是内皮细胞线粒体损伤的重要原因,而内皮细胞线粒体损伤引起细胞内信号转导异常和代谢紊乱,导致细胞的严重损伤[5]。本文重点阐述动脉粥样硬化病理进程中线粒体氧化应激引起内皮细胞损伤的作用及其机制。
-
内皮细胞有提供血液与组织间屏障和调节血管张力、血流动力学、炎症反应的功能,同时具有合成并释放血管调节因子的内分泌功能,参与维持血管结构和功能的完整及血管内环境的稳态[6]。血压的变化、血液剪切应力的变化、炎症反应和脂质累积等理化因素持续刺激内皮细胞时,血管内皮依赖性血管舒张反应受损、血管内皮完整性和通透性变化等损伤导致的内皮细胞功能障碍,是动脉粥样硬化病变的重要原因[7]。内皮细胞产生的活性氧(ROS)导致的内皮细胞损伤和功能障碍,在动脉粥样硬化的整体病理进程中起重要作用。ROS激活的ISR引起促炎细胞因子表达和炎症小体激活,促进白细胞(尤其是单核细胞)粘附并迁移到血管壁中,导致的炎症反应及内皮细胞的凋亡和脱落,是动脉粥样硬化早期病程进展的关键[2,8];ROS引起的内皮细胞通透性改变和屏障功能障碍,导致血液中活性物质侵入内皮细胞和血管平滑肌细胞中,刺激血管平滑肌细胞迁移、胶原沉积和纤维增生,这一过程是血管厚度增加和动脉粥样硬化斑块形成的原因;内皮细胞通透性改变导致的细胞中Ca2+释放,刺激血管平滑肌细胞钙化,加速动脉粥样硬化斑块形成[9]。此外,内皮细胞间紧密连接的损伤引起的动脉粥样硬化斑块破裂,导致血栓的形成、血管堵塞,在整体血管水平上表现为管腔直径、管壁厚度的改变[10]。
-
氧化应激是细胞内氧化与抗氧化作用失衡的一种状态。线粒体电子传递链的复合体I(NADH-CoQ还原酶)、复合体III(细胞色素c还原酶)泄漏未正确传递的电子至线粒体基质,而电子与O2反应产生超氧阴离子是线粒体ROS(mtROS)产生的主要原因[11]。超氧化物歧化酶(SOD)1型、2型和过氧化氢酶(CAT)以及谷胱甘肽过氧化物酶(GPx)构成抗氧化酶系统,抗氧化酶系统清除ROS的过程是细胞内的主要抗氧化途径。mtROS产生与清除的动态平衡是细胞维持线粒体稳态的重要机制,这种动态平衡被打破导致的线粒体损伤称为线粒体氧化应激[12]。
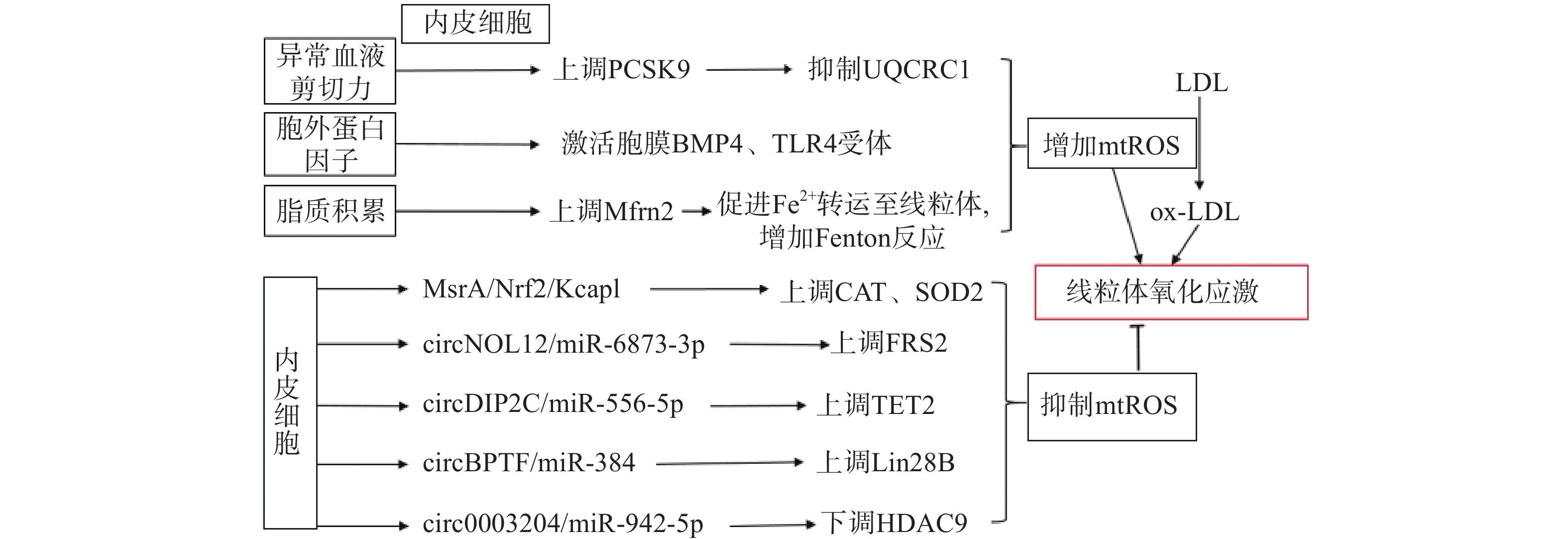
生理状态下,内皮细胞的线粒体电子传递链将大部分电子传递至氧化磷酸化产生ATP,这一过程中,仅产生少量的mtROS作为信号分子调节细胞的功能[13]。但动脉粥样硬化相关病理因素通过诱导内皮细胞mtROS的大量产生,导致线粒体氧化应激(图1)。异常的血流剪切力上调血脂代谢相关前蛋白转化酶枯草溶菌素9(PCSK9)的表达,抑制呼吸链复合体III的亚基泛醇-细胞色素c还原酶核心蛋白1(UQCRC1)的表达,导致线粒体呼吸产生的电子不能正常传递,大量泄漏至线粒基质,诱导大量mtROS的产生[14]。胞外蛋白因子通过激活内皮细胞膜上的骨形态发生蛋白4(BMP4)和Toll样受体4(TLR4)增加mtROS的产生[15]。动脉粥样硬化的重要病因之一是脂质代谢异常,而大量脂质聚集在内皮细胞时,线粒体内膜中铁转运蛋白2(Mfrn2)的表达增加,Mfrn2将胞质中Fe2+转运至线粒体参与Fenton反应,增加mtROS的产生[16];同时,脂质中的低密度脂蛋白(LDL)被mtROS氧化修饰形成氧化型低密度脂蛋白(ox-LDL)并进一步累积在内皮细胞导致大量mtROS的产生,形成的恶性循环导致线粒体氧化应激[17]。
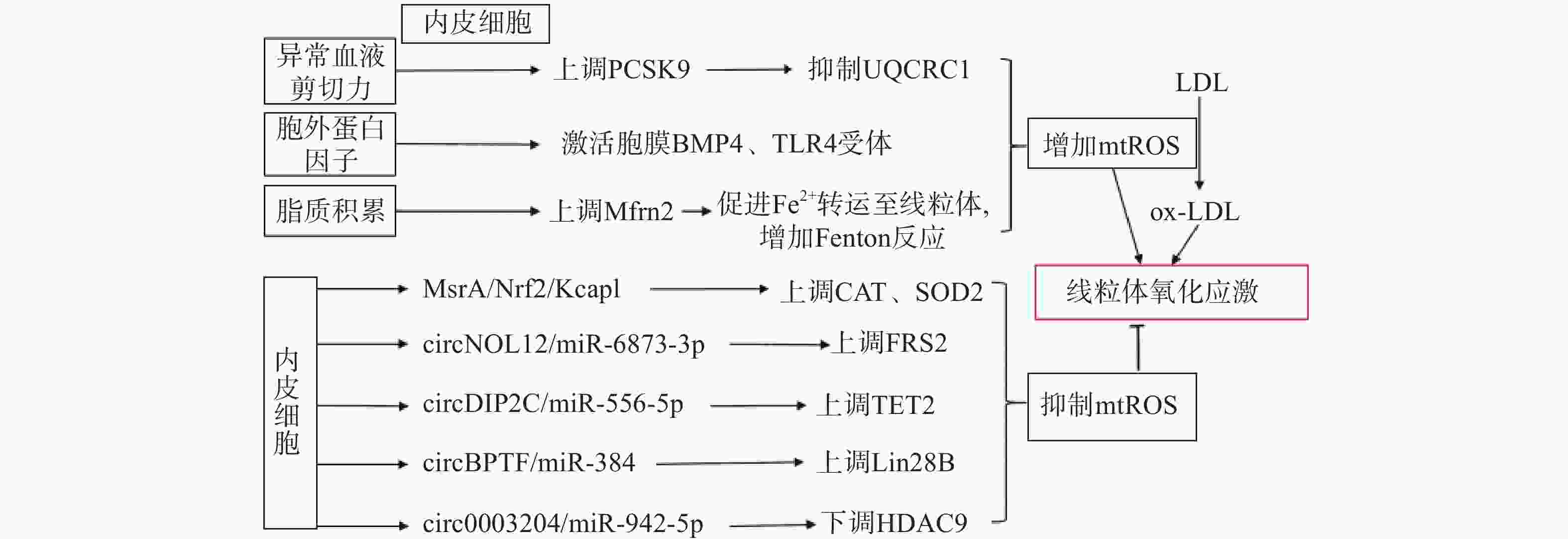
图 1 内皮细胞调控线粒体氧化应激的通路示意图
内皮细胞内的一些分子信号通路,通过直接减少mtROS的产生或者上调抗氧化酶活性间接减少mtROS,防止线粒体氧化应激,维持细胞稳态(如图1)。研究发现,甲硫氨酸被氧化时,甲硫氨酸亚砜还原酶A(MsrA)的激活,通过增加核因子E2相关因子2(Nrf2)的磷酸化,抑制Kelch样环氧氯丙烷相关蛋白1(Keap1)的表达,上调CAT、SOD2的活性[18]。而大量脂质聚集在内皮细胞时,内皮细胞部分稳态调控机制激活,其中环状RNA(circRNA)通过调节microRNA(miRNA)与mRNA相应位点的结合,实现对氧化和抗氧化相关蛋白质的调控,达到抑制mtROS产生的作用,是新的研究热点。内皮细胞的circNOL12通过竞争性抑制miR-6873-3p,上调成纤维生长因子受体底物2(FRS2)的表达,抑制mtROS的产生[19];内皮细胞的circDIP2C通过竞争性抑制miR-556-5p,上调甲基胞嘧啶双加氧酶2(TET2)的表达,抑制mtROS的产生[20];内皮细胞的circ0003204通过竞争性抑制miR-942-5p,下调诱导mtROS产生的组蛋白去乙酰化酶9(HDAC9)的表达[21];内皮细胞的circBPTF通过竞争性抑制miR-384,上调保守RNA结合蛋白LIN-28同源物B(Lin28B)的表达,抑制mtROS的产生[22]。由此可见,内皮细胞受病理因素刺激时,通过复杂的信号调节网络维持mtROS产生与清除的动态平衡,防止mtROS的过量产生,达到调控线粒体氧化应激的目的。
-
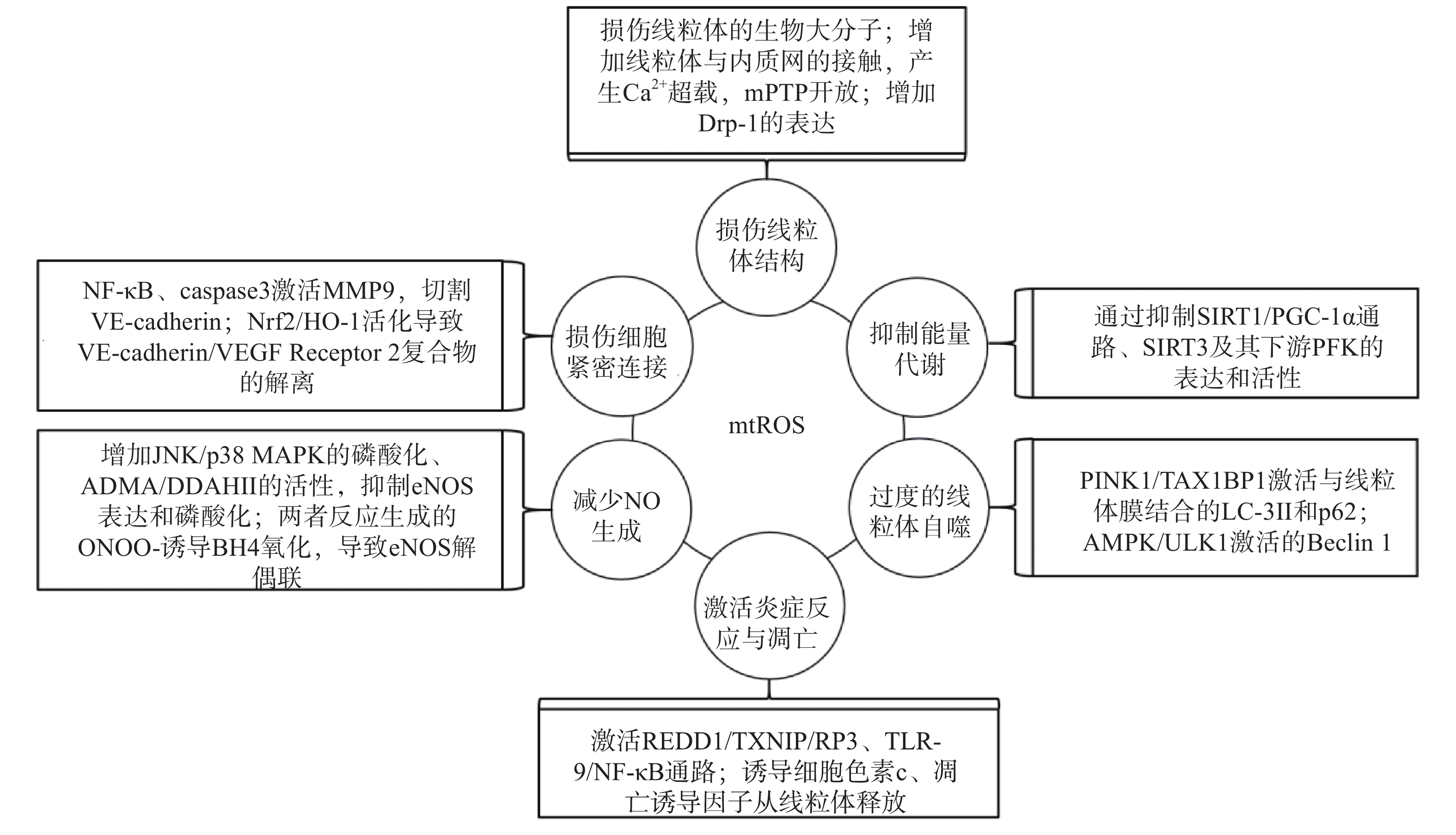
研究表明,内皮细胞的线粒体氧化应激直接引起线粒体结构和功能的严重损伤,进而导致细胞的损伤甚至死亡;而线粒体氧化应激中释放到胞浆的ROS通过诱导能量代谢异常、线粒体自噬、炎症反应、细胞凋亡、一氧化氮减少和细胞间紧密连接的损伤,形成复杂的作用网络(图2),导致内皮细胞功能的损伤,同时增加内皮细胞的代谢需求,导致线粒体损伤的持续积累。
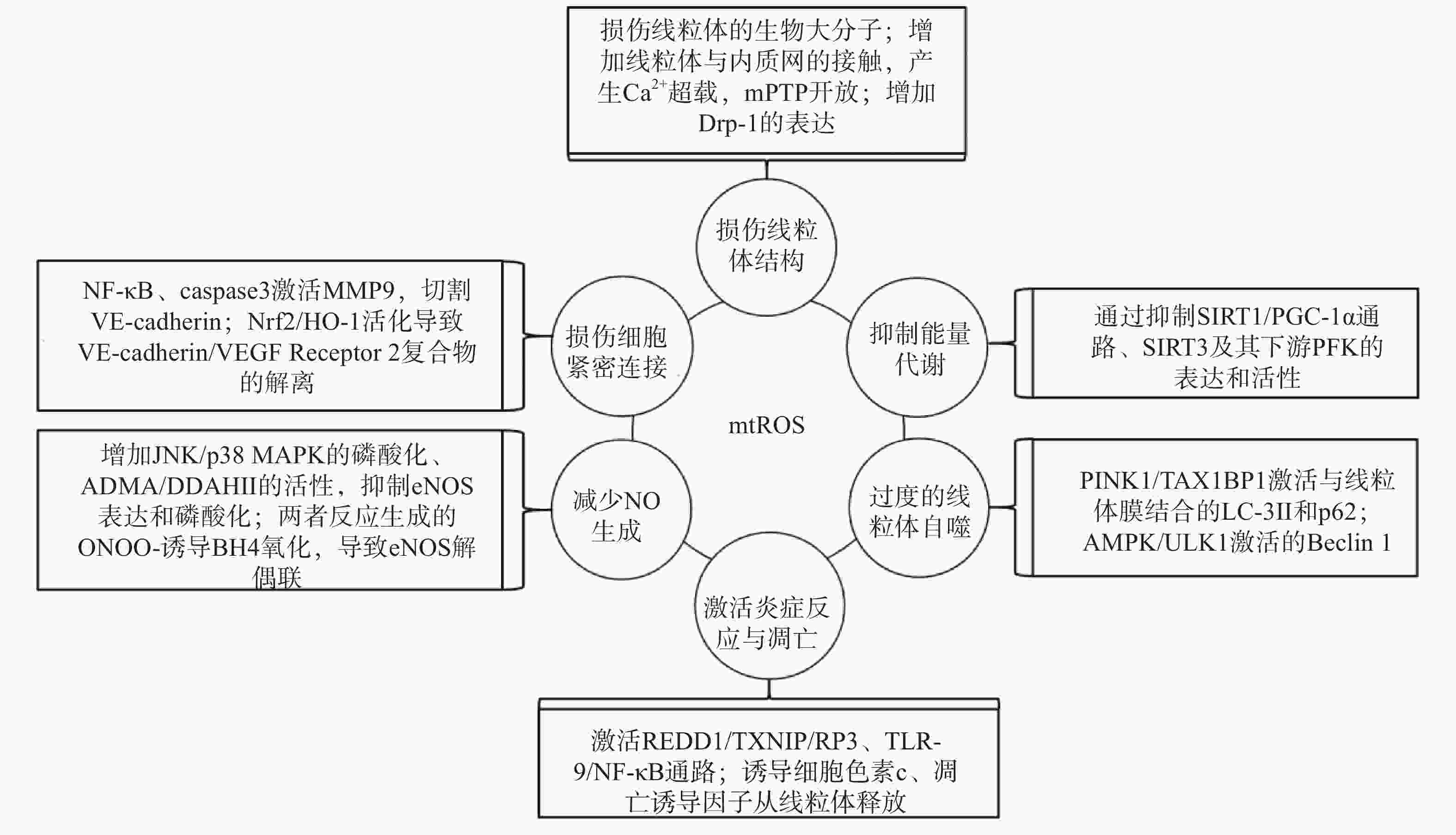
图 2 mtROS损伤内皮细胞机制的总体图
-
线粒体中的生物大分子容易受到mtROS的氧化损伤。线粒体磷脂膜被mtROS氧化时,流动性降低及通透性增加,导致线粒体膜电位丧失,线粒体失去完整性,mtROS及其他内容物释放到胞质中[23];线粒体呼吸链中的蛋白质组分被ROS氧化时,导致氧化磷酸化减少,引起线粒体的功能障碍[24];ROS引起缺少组蛋白保护的线粒体DNA(mtDNA)链断裂和突变的增加,减少线粒体功能性蛋白质的表达及线粒体相关的分子信号的变化,导致线粒体功能障碍[25]。研究还发现,mtROS能上调调节线粒体相关内质网膜形成的锚定蛋白磷酸尿苷酸性簇分类蛋白2(PACS2)的表达,通过增加线粒体与内质网的接触引起内质网内Ca2+持续性向线粒体的转运,导致线粒体渗透性转换孔(mPTP)开放、线粒体膜电位的下降,线粒体失去结构完整性[26]。有研究发现,mtROS能上调参与线粒体分裂的动力相关蛋白-1(Drp-1)的表达,导致线粒体由细长杆形、高度互通的网状结构变成小尺寸椭圆形,嵴变得不明显,线粒体整体结构受到破坏[27]。
-
能量代谢将化合物中的能量转移至ATP,为内皮细胞的生理活动提供能量储备。内皮细胞ATP的80%~85 %来自糖酵解,15%~20 %来自线粒体氧化磷酸化。mtROS通过直接损伤参与能量代谢的生物大分子抑制细胞能量代谢,而且还通过一系列反应间接调控代谢的进行。研究发现,内皮细胞内活化的沉默信息调节因子2相关酶家族(SIRT)是一种烟酰胺腺嘌呤二核苷酸(NAD+)非依赖性脱乙酰酶,通过上调转录因子表达和酶脱乙酰化,在细胞能量代谢中发挥关键作用[28]。SIRT3参与了线粒体ATP的生成、电子传递、调节线粒体呼吸链和去乙酰化激活SOD2等线粒体的大多数生物过程。同时有研究表明,SIRT3通过调节磷酸果糖激酶(PFK)的表达和活性,在内皮细胞糖酵解中发挥重要作用[29],而过度产生的mtROS会降低SIRT3的表达和活性,抑制细胞的能量代谢[30]。SIRT1通过正向调节过氧化物酶体增殖激活受体γ共激活因子1α(PGC-1α)的表达和活性,维持线粒体膜电位、线粒体动力学和氧化磷酸化的稳定,而过度产生的mtROS通过影响SIRT1/PGC-1α信号通路,抑制线粒体能量代谢[31]。内皮细胞能量代谢受抑制时,ATP生成减少导致内皮细胞生理活动的减少,引起内皮细胞的严重损伤和功能障碍。
-
自噬是细胞内通过溶酶体降解受损细胞器的过程。线粒体氧化应激能够通过内皮细胞的相关信号通路,启动和调节细胞内的线粒体自噬,清除受损线粒体,维持细胞稳态。研究表明,mtROS降低线粒体膜电位,导致PTEN诱导激酶1(PINK1)无法进入线粒体内膜与目标蛋白结合,而线粒体外膜稳定存在的PINK1通过多种调节途径引起线粒体自噬:激活并促进Tax1结合蛋白1(TAX1BP1)与细胞内自噬相关因子微管相关蛋白1轻链3 Ⅱ型(LC-3Ⅱ)的结合,诱导自噬小体定向吞噬线粒体[32];磷酸化激活E3泛素连接酶parkin(也称为p62),诱导自噬小体膜与线粒体膜的结合[33]。PINK1受mtROS影响大量存在于线粒体外膜时,生理调节途径异常活化,导致线粒体自噬的过度激活,不止受损线粒体,功能正常的线粒体同样被自噬清除,同时激活自噬相关途径的细胞死亡,引发内皮细胞的严重损伤和功能障碍[32-34]。另有研究表明,mtROS通过激活腺苷酸活化蛋白激酶(AMPK)/unc-51样自噬激活激酶1(ULK1)轴,激活Beclin 1介导的内皮细胞线粒体自噬。在这一过程中,敲除细胞内凝集素样氧化低密度脂蛋白受体-1(LOX-1),会抑制细胞内的线粒体自噬,提示LOX-1有促进线粒体自噬的作用[35]。
-
细胞凋亡又称程序性死亡,是基因控制的细胞自主有序的死亡方式。炎症反应是血管系统应对损伤因子刺激时引起的以防御为主的反应,是损伤修复的过程。动脉粥样硬化病理过程中,内皮细胞是炎症反应发生时最先受损的部位,而且研究发现内皮细胞线粒体氧化应激同时导致炎症反应和细胞凋亡[25]。受mtROS激活的DNA损伤反应调节因子1(REDD1)作为炎症始动因子,激活下游硫氧还原蛋白相互作用蛋白(TXNIP),TXNIP是一种ROS敏感蛋白质,可以直接与核苷酸结合寡聚化结构域样受体蛋白3(NLRP3)炎症小体结合并促进其活化[36],受mtROS激活的ISR信号也参与NLRP3的激活[2];NLRP3通过募集并激活凋亡因子caspase和促炎细胞因子IL-1β、IL-18等,诱导细胞炎症反应和凋亡,同时抑制SOD2的活性,导致严重的细胞损伤[37-38]。mtROS引起mtDNA泄露到胞浆,激活细胞内识别未甲基化CpG二核苷酸的toll样受体9(TLR-9),TLR-9诱导核转录因子Kappa-B(NF-κB)的激活和核转移,导致炎症反应的发生[39];mtROS激活线粒体内膜的单胺氧化酶A型(MAO-A),MAO-A催化5-HT降解的过程,也参与TLR-9的激活[40]。mtROS导致内质网内Ca2+泄露到胞浆并激活钙蛋白酶1(calpain-1),calpain-1诱导细胞色素c从线粒体释放到胞浆,进入胞浆的细胞色素c激活caspase-3,同时促进促凋亡蛋白Bax的激活及移位到线粒体,导致细胞凋亡的发生[41];mtROS激活的p38丝裂原活化蛋白激酶(p38 MAPK)也参与Bax、caspase-3的激活[40]。研究发现,mtROS还通过直接诱导线粒体内细胞色素c、凋亡诱导因子(AIF)向胞浆释放,引发炎症反应,同时激活线粒体途径凋亡因子caspase-9,导致细胞凋亡[42-43]。炎症反应引起内皮细胞的凋亡,从整个血管内皮结构上脱落,导致内皮细胞屏障功能和调节功能的失效异常。
-
内皮细胞合成和释放的一氧化氮(NO)是血管内皮依赖性舒张功能的主要调节因子。此外,NO还具有抑制血小板聚集和粘附、预防血栓形成、调节血管平滑肌细胞增殖的功能[44]。内皮细胞中NO的合成依赖于偶联形式的内皮型一氧化氮合酶(eNOS),mtROS能显著降低eNOS表达和磷酸化,减少NO的生成[45]。此外,mtROS能够与NO迅速反应生成过氧亚硝基阴离子(ONOO-)消耗已生成的NO,而且这一反应通过竞争性抑制SOD2与mtROS的反应过程拮抗SOD2的活性,导致mtROS的清除减少,产生恶性循环持续降低内皮细胞的NO。而ONOO-诱导作为eNOS辅助因子的四氢生物蝶呤(BH4)氧化,导致eNOS解偶联并转化为促氧化剂,反过来刺激ROS的产生,对细胞造成损伤[46]。mtROS还能通过多种通路间接影响NO的生成,激活C-Jun氨基末端激酶(JNK)/ p38 MAPK通路的磷酸化,抑制eNOS表达与活性[47];降低非对称性二甲基精氨酸(ADMA)的表达,增加下游二甲基精氨酸二甲胺水解酶Ⅱ(DDAH Ⅱ)的表达和活性,抑制eNOS磷酸化[48];抑制烟酰胺核苷酸转氢酶(NNT)活性,抑制eNOS的磷酸化[49]。NO浓度降低,引起的血管内皮舒张功能障碍,加重血液剪切应力引起的内皮细胞结构和功能的损伤;还加速血小板的聚集和血管平滑肌细胞的增殖,促进了动脉粥样硬化斑块的形成。
-
mtROS导致细胞间紧密连接异常是血管内皮屏障功能障碍的主要原因之一。研究发现,mtROS激活的NF-κB诱导炎症反应的同时,还诱导基质金属蛋白酶9(MMP9)的表达,mtROS经细胞色素c途径激活的caspase也能诱导MMP9的表达,而MMP9切割VE-钙粘蛋白(VE-cadherin),破坏细胞间紧密连接[50]。还有研究发现,mtROS促进Nrf2从细胞质移位到细胞核,上调血红素加氧酶1(HO-1)的表达,HO-1能上调抗氧化酶活性,同时还通过诱导血管内皮生长因子A(VEGF-A,也称为血管通透性因子)的分泌,引起细胞膜上VE-cadherin /VEGF Receptor 2复合物的解离,导致细胞间紧密连接的不连续[51]。内皮细胞间紧密连接的损伤,在动脉粥样硬化早期导致血液中物质侵入血管平滑肌细胞,引发炎症和组织钙化;在斑块形成之后,内皮细胞间紧密连接的损伤导致斑块的不稳定和破裂,导致血栓的形成。
-
综上所述,线粒体氧化应激作为初始损伤因素,通过多种机制的共同作用诱导炎症反应、细胞凋亡、过度自噬等损伤事件,导致血管内皮功能障碍。对这一病理过程所涉及损伤事件的深入研究,越来越多的线粒体氧化应激诱导内皮细胞损伤的相关分子信号通路和内皮细胞对线粒体氧化应激的调控机制被阐明,有助于增加对线粒体氧化应激损伤内皮细胞机制的认识和理解动脉粥样硬化发病过程,并为从初始损伤因素缓解动脉粥样硬化病理进程提供有益的参考。但是深入研究线粒体氧化应激损伤内皮细胞机制的同时,针对线粒体的抗氧化研究则略显不足,目前临床使用的通过抗氧化作用改善内皮细胞氧自由基产生的药物有普罗布考和维生素,为数不多。仍需进一步研究通过抑制ROS大量产生或者激活细胞内固有抗氧化系统进而缓解线粒体氧化应激、并维持内皮细胞结构和功能完整性的治疗策略,将线粒体氧化应激作为临床治疗动脉粥样硬化的重要靶点,减少内皮细胞的初始损失并降低动脉粥样硬化患者的发病率。
Mitochondrial oxidative stress in vascular endothelial cell and atherosclerosis
-
摘要: 血管内皮细胞损伤是动脉粥样硬化病理过程的起始环节。线粒体氧化应激与血管内皮细胞功能密切相关,线粒体氧化应激通过诱导线粒体自噬、一氧化氮生成减少、炎症反应、细胞代谢失衡和凋亡,导致血管内皮细胞的功能障碍。同时,血管内皮细胞也通过调控线粒体氧化应激维持自身稳态。本文旨在综述动脉粥样硬化病理过程中线粒体氧化应激诱发血管内皮细胞损伤的主要分子信号通路,为后续研究两者间的分子机制提供参考。Abstract: The injury of vascular endothelial cell function is the beginning of the pathological process of atherosclerosis. Mitochondrial oxidative stress is closely related to vascular endothelial cell function, which causes the dysfunction of vascular endothelial cell by inducing mitophagy, reducing nitric oxide production, inflammation, cellular metabolic imbalance and apoptosis. Meanwhile, vascular endothelial cell could also maintain their homeostasis by regulating mitochondrial oxidative stress. The molecular signaling pathways of the vascular endothelial cell injury caused by mitochondrial oxidative stress in the pathological process of atherosclerosis were outlined in this review, which provided reference for further research on the molecular mechanism between mitochondrial oxidative stress and endothelial damage.
-
[1] KAI H H, WU Q Y, YIN R H, et al. LncRNA NORAD promotes vascular endothelial cell injury and atherosclerosis through suppressing VEGF gene transcription via enhancing H3K9 deacetylation by recruiting HDAC6[J]. Front Cell Dev Biol,2021,9:701628. doi: 10.3389/fcell.2021.701628 [2] FUSTER J J. Integrated stress response inhibition in atherosclerosis[J]. J Am Coll Cardiol,2019,73(10):1170-1172. doi: 10.1016/j.jacc.2019.01.015 [3] ZENG J F, TAO J, XIA L Z, et al. Melatonin inhibits vascular endothelial cell pyroptosis by improving mitochondrial function via up-regulation and demethylation of UQCRC1[J]. Biochem Cell Biol,2021,99(3):339-347. doi: 10.1139/bcb-2020-0279 [4] KLUGE M A, FETTERMAN J L, VITA J A. Mitochondria and endothelial function[J]. Circ Res,2013,112(8):1171-1188. doi: 10.1161/CIRCRESAHA.111.300233 [5] DYMKOWSKA D. The involvement of autophagy in the maintenance of endothelial homeostasis: The role of mitochondria[J]. Mitochondrion,2021,57:131-147. doi: 10.1016/j.mito.2020.12.013 [6] YAO G H, QI J J, ZHANG Z Y, et al. Endothelial cell injury is involved in atherosclerosis and lupus symptoms in gld. apoE-/- mice[J]. Int J Rheum Dis,2019,22(3):488-496. doi: 10.1111/1756-185X.13458 [7] ZHANG X H, LU J Y, ZHANG Q H, et al. CircRNA RSF1 regulated ox-LDL induced vascular endothelial cells proliferation, apoptosis and inflammation through modulating miR-135b-5p/HDAC1 axis in atherosclerosis[J]. Biol Res,2021,54(1):11. doi: 10.1186/s40659-021-00335-5 [8] LIU X Y, XU Y L, CHENG S B, et al. Geniposide combined with notoginsenoside r1 attenuates inflammation and apoptosis in atherosclerosis via the AMPK/mTOR/Nrf2 signaling pathway[J]. Front Pharmacol,2021,12:687394. doi: 10.3389/fphar.2021.687394 [9] BIAN W H, JING X H, YANG Z Y, et al. Downregulation of LncRNA NORAD promotes Ox-LDL-induced vascular endothelial cell injury and atherosclerosis[J]. Aging,2020,12(7):6385-6400. doi: 10.18632/aging.103034 [10] MA Y Y, LIANG X R, LI C, et al. 5-HT 2A receptor and 5-HT degradation play a crucial role in atherosclerosis by modulating macrophage foam cell formation, vascular endothelial cell inflammation, and hepatic steatosis[J]. J Atheroscler Thromb,2022,29(3):322-336. doi: 10.5551/jat.58305 [11] DIKALOV S, ITANI H, RICHMOND B, et al. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension[J]. Am J Physiol Heart Circ Physiol,2019,316(3):H639-H646. doi: 10.1152/ajpheart.00595.2018 [12] JIANG W, GENG H Z, LV X Q, et al. Idebenone protects against atherosclerosis in apolipoprotein E-Deficient mice via activation of the SIRT3-SOD2-mtROS pathway[J]. Cardiovasc Drugs Ther,2021,35(6):1129-1145. doi: 10.1007/s10557-020-07018-5 [13] KATTOOR A J, POTHINENI N V K, PALAGIRI D, et al. Oxidative stress in atherosclerosis[J]. Curr Atheroscler Rep,2017,19(11):42. doi: 10.1007/s11883-017-0678-6 [14] ZENG J F, TAO J, XI L Z, et al. PCSK9 mediates the oxidative low-density lipoprotein-induced pyroptosis of vascular endothelial cells via the UQCRC1/ROS pathway[J]. Int J Mol Med,2021,47(4):53. doi: 10.3892/ijmm.2021.4886 [15] CHOY K W, LAU Y S, MURUGAN D, et al. Paeonol attenuates LPS-Induced endothelial dysfunction and apoptosis by inhibiting BMP4 and TLR4 signaling simultaneously but independently[J]. J Pharmacol Exp Ther,2018,364(3):420-432. doi: 10.1124/jpet.117.245217 [16] WANG D C, YE P, KONG C H, et al. Mitoferrin 2 deficiency prevents mitochondrial iron overload-induced endothelial injury and alleviates atherosclerosis[J]. Exp Cell Res,2021,402(1):112552. doi: 10.1016/j.yexcr.2021.112552 [17] CHEN L, HU L Q, LI Q, et al. Exosome-encapsulated miR-505 from ox-LDL-treated vascular endothelial cells aggravates atherosclerosis by inducing NET formation[J]. Acta Biochim Biophys Sin (Shanghai),2019,51(12):1233-1241. doi: 10.1093/abbs/gmz123 [18] WU Y, SONG F, LI Y D, et al. Acacetin exerts antioxidant potential against atherosclerosis through Nrf2 pathway in apoE-/ - Mice[J]. J Cell Mol Med,2021,25(1):521-534. doi: 10.1111/jcmm.16106 [19] LI S Z, HAO M H, WU T S, et al. Kaempferol alleviates human endothelial cell injury through circNOL12/miR-6873-3p/FRS2 axis[J]. Biomed Pharmacother,2021,137:111419. doi: 10.1016/j.biopha.2021.111419 [20] HU F D, CHEN X, GAO J, et al. CircDIP2C ameliorates oxidized low-density lipoprotein-induced cell dysfunction by binding to miR-556-5p to induce TET2 in human umbilical vein endothelial cells[J]. Vascul Pharmacol,2021,139:106887. doi: 10.1016/j.vph.2021.106887 [21] WAN H, YOU T, LUO W. Circ_0003204 Regulates Cell Growth, Oxidative Stress, and Inflammation in ox-LDL-Induced Vascular Endothelial Cells via Regulating miR-942-5p/HDAC9 Axis[J]. Front Cardiovasc Med,2021,8:646832. doi: 10.3389/fcvm.2021.646832 [22] ZHANG W, SUI Y. CircBPTF knockdown ameliorates high glucose-induced inflammatory injuries and oxidative stress by targeting the miR-384/LIN28B axis in human umbilical vein endothelial cells[J]. Mol Cell Biochem,2020,471(1-2):101-111. doi: 10.1007/s11010-020-03770-2 [23] MARCHIO P, GUERRA-OJEDA S, VILA J M, et al. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation[J]. Oxidative Med Cell Longev,2019,2019:8563845. [24] FILOMENI G, De ZIO D, CECCONI F. Oxidative stress and autophagy: The clash between damage and metabolic needs[J]. Cell Death Differ,2015,22(3):377-388. doi: 10.1038/cdd.2014.150 [25] PATERGNANI S, BOUHAMIDA E, LEO S, et al. Mitochondrial oxidative stress and “mito-inflammation”: Actors in the diseases[J]. Biomedicines,2021,9(2):216. doi: 10.3390/biomedicines9020216 [26] YU S J, ZHANG L P, LIU C, et al. PACS2 is required for ox-LDL-induced endothelial cell apoptosis by regulating mitochondria-associated ER membrane formation and mitochondrial Ca2 + elevation[J]. Exp Cell Res,2019,379(2):191-202. doi: 10.1016/j.yexcr.2019.04.002 [27] HUANG M J, WEI R B, WANG Y, et al. The uremic toxin hippurate promotes endothelial dysfunction via the activation of Drp1-mediated mitochondrial fission[J]. Redox Biol,2018,16:303-313. doi: 10.1016/j.redox.2018.03.010 [28] MAIESE K. Harnessing the power of SIRT1 and non-coding RNAs in vascular disease[J]. Curr Neurovasc Res,2017,14(1):82-88. doi: 10.2174/1567202613666161129112822 [29] HE X C, ZENG H, CHEN S T, et al. Endothelial specific SIRT3 deletion impairs glycolysis and angiogenesis and causes diastolic dysfunction[J]. J Mol Cell Cardiol,2017,112:104-113. doi: 10.1016/j.yjmcc.2017.09.007 [30] WU J, DENG Z Y, SUN M M, et al. Polydatin protects against lipopolysaccharide-induced endothelial barrier disruption via SIRT3 activation[J]. Lab Invest,2020,100(4):643-656. doi: 10.1038/s41374-019-0332-8 [31] TSAI K L, HUNG C H, CHAN S H, et al. Chlorogenic acid protects against oxLDL-Induced oxidative damage and mitochondrial dysfunction by modulating SIRT1 in endothelial cells[J]. Mol Nutr Food Res,2018,62(11):e1700928. doi: 10.1002/mnfr.201700928 [32] FAN Y Z, CHENG Z L, MAO L J, et al. PINK1/TAX1BP1-directed mitophagy attenuates vascular endothelial injury induced by copper oxide nanoparticles[J]. J Nanobiotechnol,2022,20(1):149. doi: 10.1186/s12951-022-01338-4 [33] NING R H, LI Y, DU Z, et al. The mitochondria-targeted antioxidant MitoQ attenuated PM 2.5-induced vascular fibrosis via regulating mitophagy[J]. Redox Biol,2021,46:102113. doi: 10.1016/j.redox.2021.102113 [34] WANG W, WU Q H, SUI Y, et al. Rutin protects endothelial dysfunction by disturbing Nox4 and ROS-sensitive NLRP3 inflammasome[J]. Biomed Pharmacother,2017,86:32-40. doi: 10.1016/j.biopha.2016.11.134 [35] QIN X, ZHANG J, WANG B, et al. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells[J]. Autophagy,2021,17(12):4266-4285. doi: 10.1080/15548627.2021.1911016 [36] HOU X H, YANG S B, YIN J. Blocking the REDD1/TXNIP axis ameliorates LPS-induced vascular endothelial cell injury through repressing oxidative stress and apoptosis[J]. Am J Physiol Cell Physiol,2019,316(1):C104-C110. doi: 10.1152/ajpcell.00313.2018 [37] TANG Y S, ZHAO Y H, ZHONG Y, et al. Neferine inhibits LPS-ATP-induced endothelial cell pyroptosis via regulation of ROS/NLRP3/Caspase-1 signaling pathway[J]. Inflamm Res,2019,68(9):727-738. doi: 10.1007/s00011-019-01256-6 [38] INCALZA M A, D'ORIA R, NATALICCHIO A, et al. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases[J]. Vascul Pharmacol,2018,100:1-19. doi: 10.1016/j.vph.2017.05.005 [39] DING Z F, LIU S J, WANG X W, et al. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis[J]. Sci Rep,2013,3:1077. doi: 10.1038/srep01077 [40] GONG L L, LEI Y Y, LIU Y X, et al. Vaccarin prevents ox-LDL-induced HUVEC EndMT, inflammation and apoptosis by suppressing ROS/p38 MAPK signaling[J]. Am J Transl Res,2019,11(4):2140-2154. [41] CHEN H I, HU W S, HUNG M Y, et al. Protective effects of luteolin against oxidative stress and mitochondrial dysfunction in endothelial cells[J]. Nutr Metab Cardiovasc Dis,2020,30(6):1032-1043. doi: 10.1016/j.numecd.2020.02.014 [42] BAI L N, YANG J H, ZHANG H, et al. PTB domain and leucine zipper motif 1 (APPL1) inhibits myocardial ischemia/hypoxia-reperfusion injury via inactivation of apoptotic protease activating factor-1 (APAF-1)/Caspase9 signaling pathway[J]. Bioengineered,2021,12(1):4385-4396. doi: 10.1080/21655979.2021.1954841 [43] KONG D X, HAN Y T, WANG C B, et al. Cytoprotective effects of oleanolic acid in human umbilical vascular endothelial cells is mediated via UCP2/ROS/Cytochrome C/AIF pathway[J]. J Cardiovasc Pharmacol,2016,67(4):344-350. doi: 10.1097/FJC.0000000000000360 [44] FÖRSTERMANN U, XIA N, LI H G. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis[J]. Circ Res,2017,120(4):713-735. doi: 10.1161/CIRCRESAHA.116.309326 [45] HE H, WANG L, QIAO Y, et al. Doxorubicin Induces Endotheliotoxicity and Mitochondrial Dysfunction via ROS/eNOS/NO Pathway[J]. Front Pharmacol,2020,10:1531. doi: 10.3389/fphar.2019.01531 [46] AKHIGBE R, AJAYI A. The impact of reactive oxygen species in the development of cardiometabolic disorders: A review[J]. Lipids Health Dis,2021,20(1):23. doi: 10.1186/s12944-021-01435-7 [47] SHAFIQUE E, TORINA A, REICHERT K, et al. Mitochondrial redox plays a critical role in the paradoxical effects of NAPDH oxidase-derived ROS on coronary endothelium[J]. Cardiovasc Res,2017,113(2):234-246. doi: 10.1093/cvr/cvw249 [48] CHEN X P, LI H W, WANG Z Q, et al. Quercetin protects the vascular endothelium against iron overload damages via ROS/ADMA/DDAHⅡ/eNOS/NO pathway[J]. Eur J Pharmacol,2020,868:172885. doi: 10.1016/j.ejphar.2019.172885 [49] RAO K N S, SHEN X G, PARDUE S, et al. Nicotinamide nucleotide transhydrogenase (NNT) regulates mitochondrial ROS and endothelial dysfunction in response to angiotensin II[J]. Redox Biol,2020,36:101650. doi: 10.1016/j.redox.2020.101650 [50] GALKIN I I, PLETJUSHKINA O Y, ZINOVKIN R A, et al. Mitochondria-Targeted Antioxidant SkQR1 Reduces TNF-Induced Endothelial Permeability in vitro[J]. Biochemistry (Mosc),2016,81(10):1188-1197. doi: 10.1134/S0006297916100163 [51] TSENG C Y, WANG J S, CHAO M W. Causation by diesel exhaust particles of endothelial dysfunctions in cytotoxicity, pro-inflammation, permeability, and apoptosis induced by ROS generation[J]. Cardiovasc Toxicol,2017,17(4):384-392. doi: 10.1007/s12012-016-9364-0 -
 点击查看大图
点击查看大图
图(2)
计量
- 文章访问数: 3575
- HTML全文浏览量: 4749
- PDF下载量: 53
- 被引次数: 0
