

-
尽管强效且具有高度选择性的小分子抑制剂仍在不断得到开发与应用,但这类传统药物并不能直接调控细胞内目标蛋白的含量,难以靶向缺乏配体结合口袋的“难成药”靶点[1-3],且起效剂量较大,长期使用耐药性明显[4-6]。因此,随着研究的不断深入,利用细胞内固有的蛋白降解机制靶向降解特定蛋白的方式,引起了科学界的极大关注,并成为一种颇具前景的治疗手段。
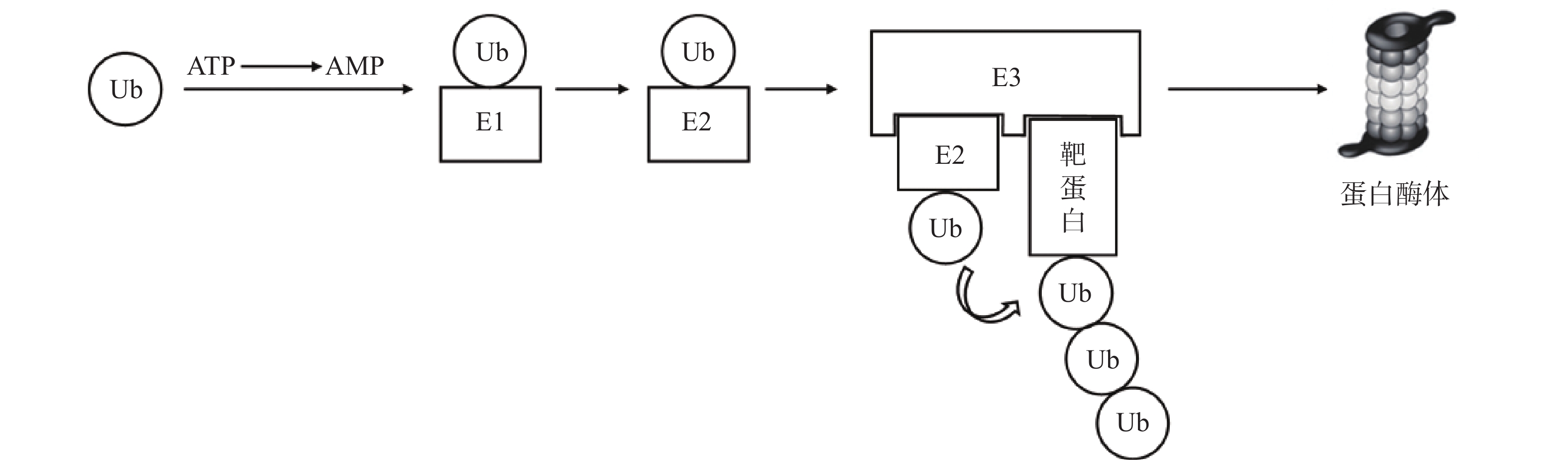
泛素-蛋白酶体系统(UPS)是细胞内降解受损或错误折叠蛋白质的核心机制,参与胞内80%以上蛋白质的降解[7]。其主要过程为:首先,泛素(Ub)与泛素激活酶(E1)形成硫酯键;活化的泛素通过硫酯交换反应被转移至泛素结合酶(E2);随后,泛素连接酶(E3)同时结合“E2-泛素复合物”与靶蛋白,使两者在空间上充分接近,驱动泛素转移至靶蛋白。该过程由ATP供能,多次重复可使底物被泛素链标记,最终被蛋白酶体识别并降解[8-9](图1)。基于UPS的靶向蛋白降解技术中,研究最为成熟的包括PROTAC和分子胶。

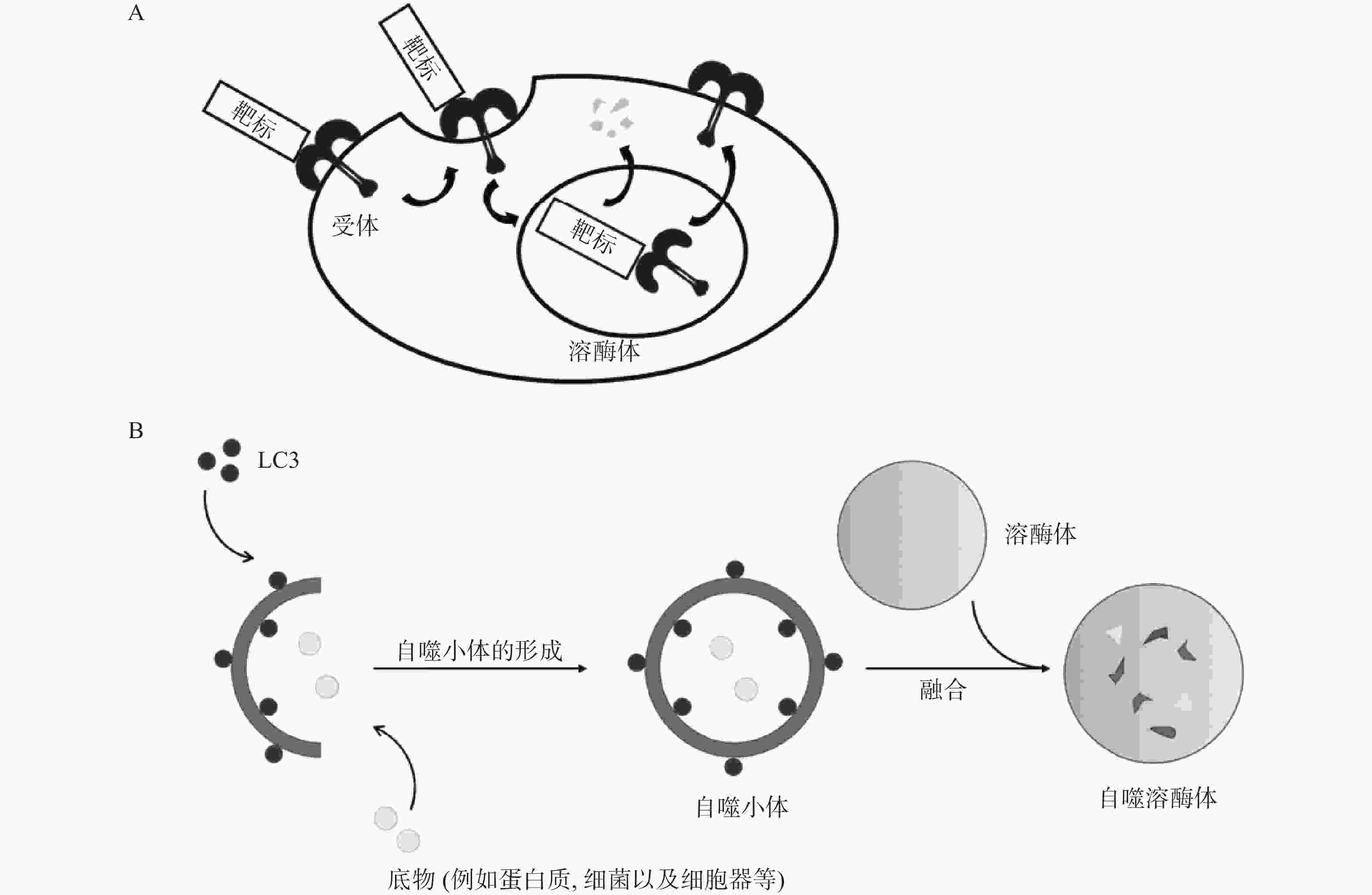
除UPS外,在真核生物中还普遍存在另一种降解途径,即溶酶体介导的降解途径。该途径涉及的底物范围十分广泛,包括衰老或受损的细胞器、蛋白质聚集体以及入侵的病原微生物等,与UPS互为补充。溶酶体降解途径主要包括内体-溶酶体途径(ELP)[10]和自噬-溶酶体途径(ALP)[11-12],因此,基于ELP的靶向降解技术主要应用于胞外蛋白和膜蛋白的靶向降解,例如LYTAC;ELP是指细胞通过受体介导的内吞作用将胞外物质或膜蛋白与内体融合,并最终转运至溶酶体中降解的过程(图2A);ALP通常是指在自噬关键蛋白LC3的参与下,通过形成自噬小体将底物包裹,最终与溶酶体融合实现降解的过程(图2B)。基于ALP的靶向降解技术进一步拓展了靶向降解胞内蛋白(包括蛋白聚集体)的技术手段,并成功应用于降解受损细胞器和其它非蛋白生物分子,主要降解技术包括ATTEC、AUTAC和AUTOTAC等。
基于以上两种途径的靶向蛋白降解技术,利用“事件驱动”(event driven)而非“占据驱动”(occupation driven)的作用模式发挥功能,理论上仅需催化量的药物即可发挥较强疗效,在靶向“难成药”靶点、提高选择性、克服耐药、降低毒副作用等方面表现出较大的优势,具有广阔的应用前景。本文综述了近年来兴起的代表性的靶向降解策略,并讨论其优势与局限性。
-
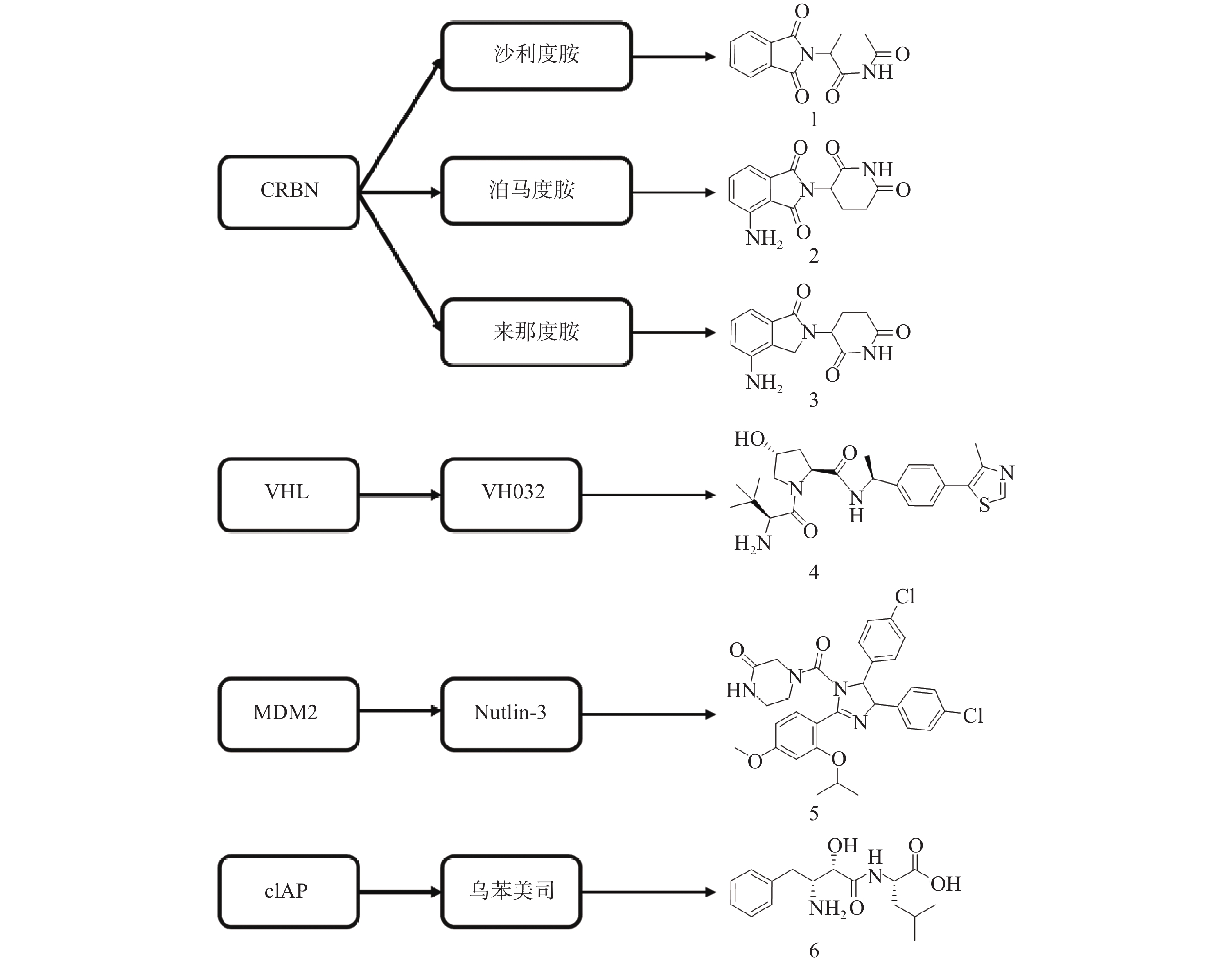
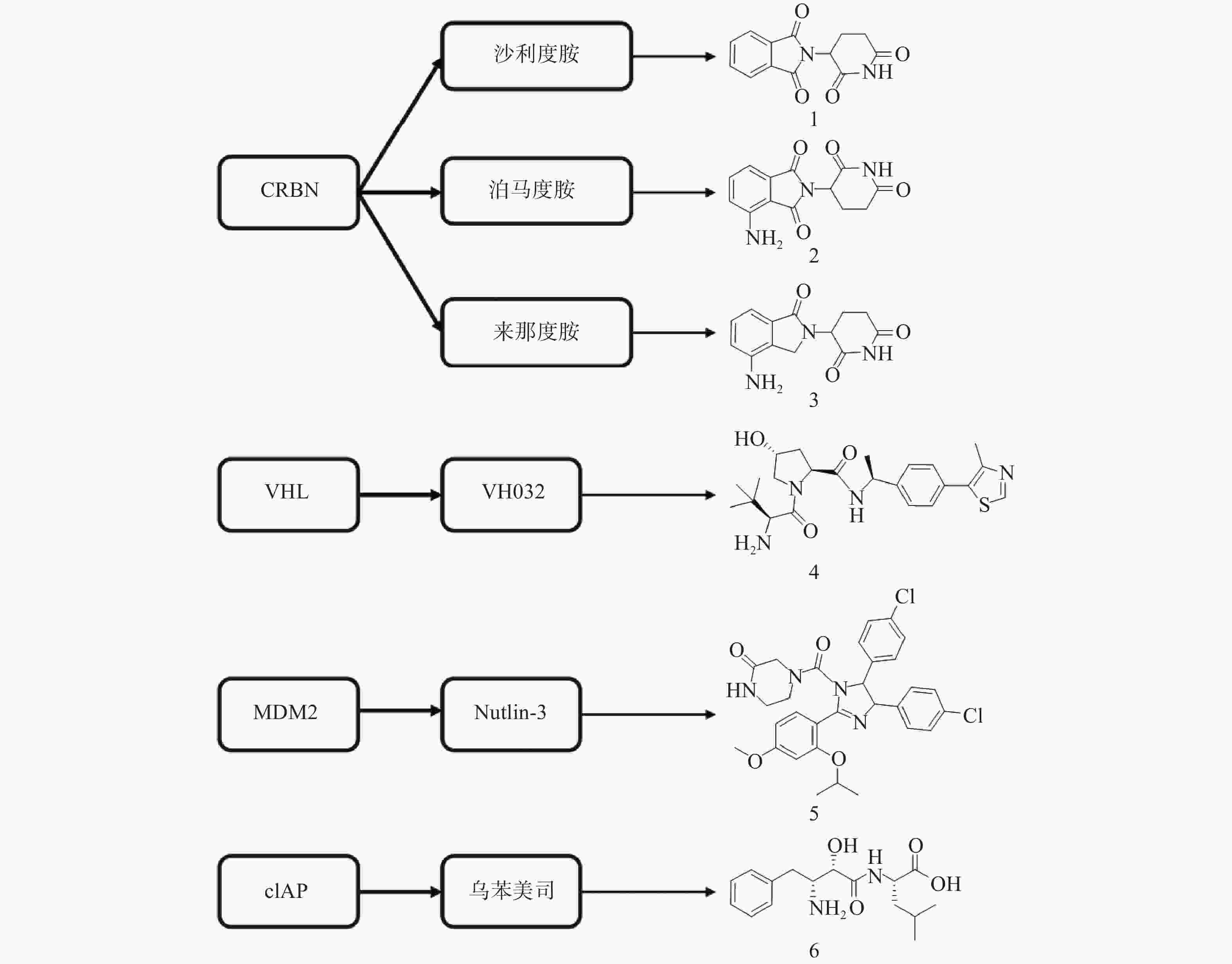
PROTAC是一类异双功能分子,一端为靶蛋白配体,另一端为E3连接酶配体,中间通过Linker相连。在细胞中PROTAC通过同时结合底物蛋白和E3,形成“靶蛋白-PROTAC分子-E3”三元复合物,诱导靶蛋白的泛素化和蛋白酶体降解。自2001年Crews等首次提出PROTAC的概念以来[13],该技术不断发展与完善,越来越多的科研人员致力于通过PROTAC靶向降解目标蛋白。目前已有多种类型靶标蛋白质可通过该策略实现降解,包括核受体(ER[14]、AR[15-27]等)、蛋白激酶(CDK4[28-29]、CDK9[30-32]、ALK[33-34]等)、蛋白转录调控蛋白(BRD4[35-40]、HDAC6[41-43]、Sirt2[44-45]等)、调节蛋白(AHR[46]等)、神经性疾病相关蛋白(Tau[47-49]等)、细胞代谢酶(MetAP2[13]等)以及融合蛋白(HaloTag[50])等。此外,尽管人类基因组可以编码600多个E3连接酶[51],但是仅有Cereblon(CRBN)、von Hippel-Lindau (VHL)、mouse double minute 2 homologue (MDM2)和cellular inhibitor of apoptosis protein-1(cIAP)等有限的几个成功用于PROTAC的构建(图3)。
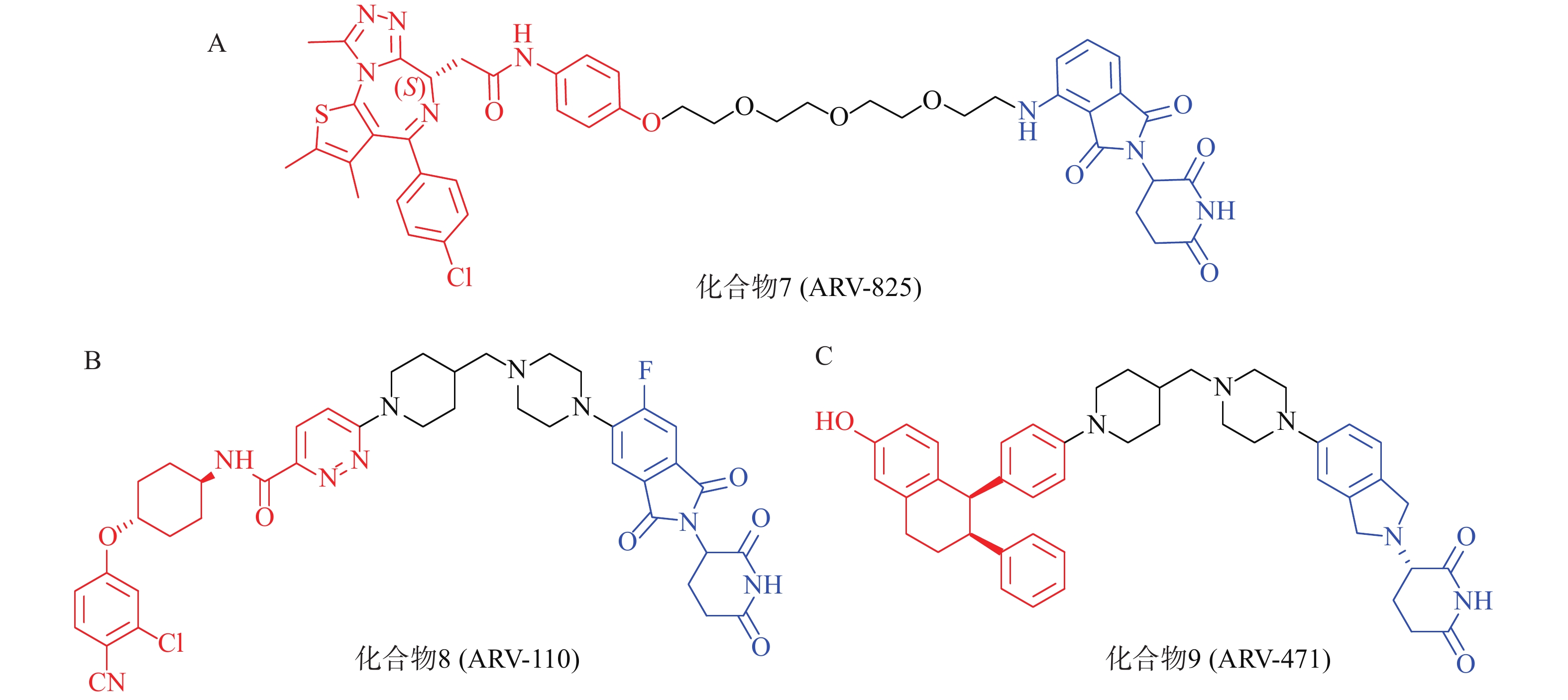
以经典靶点BRD4蛋白为例,作为BET蛋白家族中最重要的一员,BRD4在人体中广泛分布,对细胞正常生长及细胞周期的调控具有重要意义,亦是肿瘤发生的重要参与者,针对BET的抑制剂已有长期研究基础。2015年,Crews课题组设计了一种新型小分子PROTAC:ARV-825(化合物7,图4A),其一端为泊马度胺(pomalidomide),可被E3泛素连接酶CRBN识别,另一端为已进入临床试验的BRD4抑制剂OTX015,两者通过聚乙二醇链相连,用于靶向降解BRD4蛋白。Western Blot实验表明,化合物7能在BL细胞中成功诱导BRD4的高效降解(DC50<1 nmol/L)。该研究为BRD4的靶向降解提供了强有力的手段,具有深远的临床价值和治疗意义;且首次实现E3连接酶CRBN在PROTAC中的应用,为小分子PROTAC的设计奠定了基础[52]。
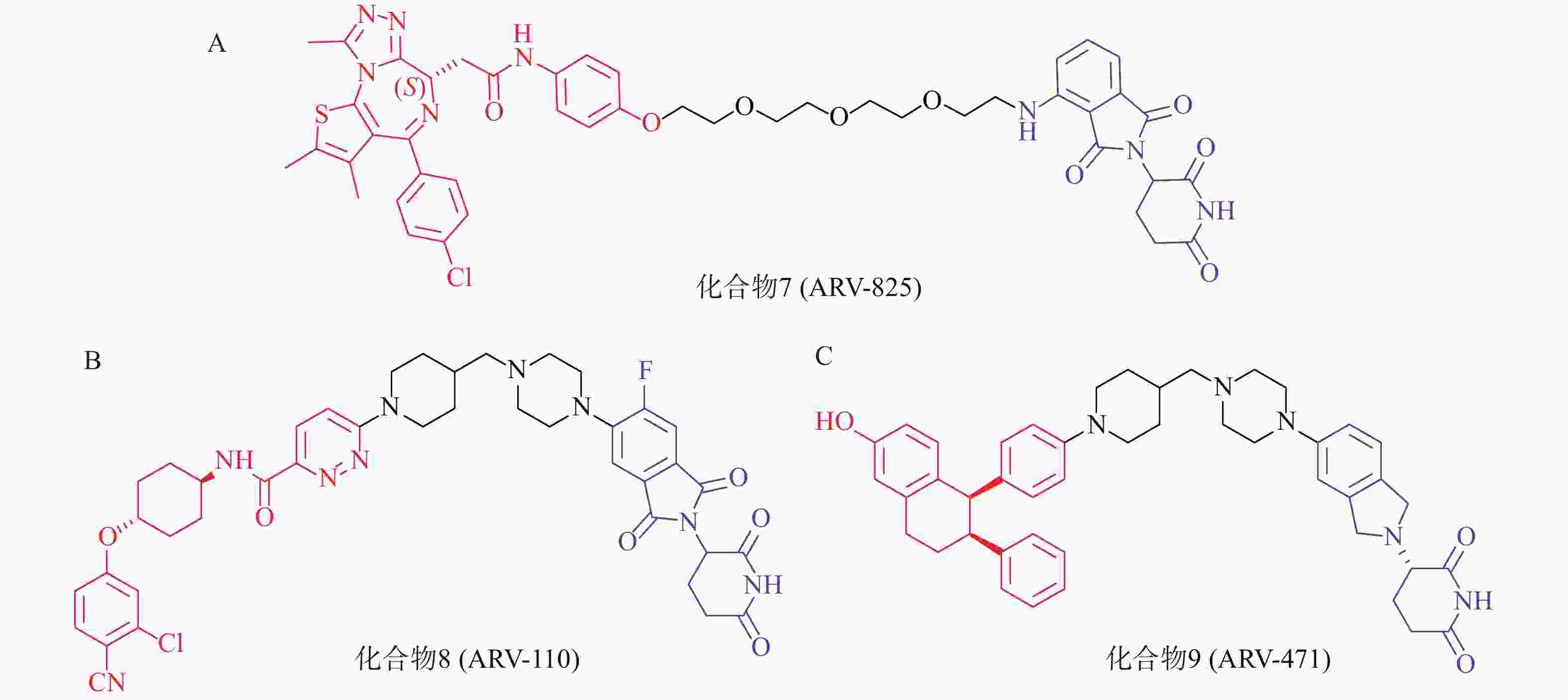
PROTAC策略正持续赋能针对多种疾病相关靶点的药物开发,当前已有约20个PROTAC分子进入临床研究阶段[53]。其中,ARV-110(化合物8,图4B)与ARV-471(化合物9,图4C)是最早进入临床研究的PROTAC药物,分别通过靶向雄激素受体(AR)与雌激素受体(ER)治疗转移性去势抵抗性前列腺癌(mCRPC)和ER+/HER2-晚期乳腺癌患者[54-56]。据Arvinas公司公布的Ⅰ期和Ⅱ临床试验数据,ARV-110对携带ART878或H875突变患者具有较强治疗作用;ARV-471能够显著降低ER+/HER2-晚期乳腺癌患者肿瘤组织中的ER表达水平,可达到38%的临床获益率(CBR)。
尽管PROTAC技术给药物开发带来了新的机遇,但也面临一些关键挑战,例如:①分子量过大、透膜性差导致的成药性问题,多数降解剂无法通过口服给药,甚至一些基于寡核苷酸或多肽的PROTAC,需要通过特殊的药物递送系统进入细胞;②E3连接酶在不同细胞和组织中的表达不同,可用于构建PROTAC的E3连接酶仅有CRBN和VHL等少数几个,针对E3连接酶配体的类药性优化难度较大;③hook effect,即PROTAC在低浓度下药效较弱,在高浓度下又因无法形成三元复合物抑制泛素化降解的过程。
-
分子胶是利用UPS实现细胞内蛋白靶向降解的另一种技术手段。分子胶通常符合“类药五原则”,与PROTAC相比具有更小的分子量(通常小于600)、更好的膜通透性和成药性,但是并不等同于“去掉连接子的PROTAC”。分子胶通过诱导或稳定E3连接酶与靶蛋白之间的“蛋白-蛋白相互作用”,介导靶蛋白泛素化和随后的蛋白酶体降解。分子胶的发现大多是偶然的,尚难以实现理性设计[57]。2014年,Ebert课题组发现免疫抑制药物Thalidomide(沙利度胺)、Pomalidomide(泊马度胺)和Lenalidomide(来那度胺)(化合物1~3,图3)在与CRBN结合后可促使锌指转录因子IKZF1和IKZF3泛素化并产生降解,进而发挥免疫调节和抗肿瘤作用[58]。后续研究发现,化合物3与CRBN的结合还可诱导酪蛋白激酶1α(CK1α)的降解[59]。在上述研究中,化合物1~3均位于E3连接酶CRBN及靶蛋白之间,充当了分子胶的作用。
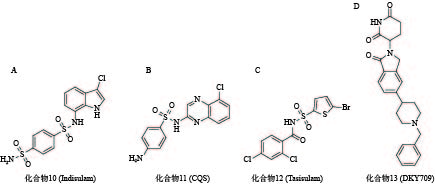
化合物10(图5A)是一种具有选择性抗肿瘤活性的芳基磺胺类药物,但其作用机制一直不明[60]。2017年,Nijhawan课题组证实化合物10在E3连接酶CUL4-DCAF15和RNA剪切因子RBM39间作为分子胶粘合剂诱导RBM39的泛素化和降解,与其结构类似的化合物11(图5B)和化合物12(图5C)也有相同机制[61]。2020年,Bussiere等解析出DCAF15-DDB1-DDA1-indisulam-RBM39的晶体结构,直观地证实了其分子胶的作用[62]。

目前已有多个分子胶进入临床研究阶段。例如,诺华公司开发的分子胶DKY709(化合物13,图5D)能够靶向降解转录因子IKZF2,正在作为单一药物,或与抗PD-1单克隆抗体联用进行I期临床试验,用于治疗晚期实体瘤患者;BioTheryX公司研发的分子胶BTX-1188(结构尚未披露)被FDA批准开展I期临床试验,通过降解GSPT1、IKZF1/3等多种蛋白质,实现对白血病及实体瘤的治疗作用。
尽管基于UPS 的靶向蛋白降解技术已经在新药研发领域得到了广泛应用,但受限于UPS的细胞内定位以及蛋白酶体狭窄的内径,UPS仅能降解具有胞质结构域的单体蛋白,对于胞外蛋白、蛋白聚集体及非蛋白物质无能为力。因此亟待拓展新的降解途径,为靶向蛋白的降解提供更多技术手段。
-
基于溶酶体途径有望解决PROTAC等技术的瓶颈问题,发挥协同或补偿作用。其中依赖ELP的策略包括LYTAC,依赖ALP的策略包括ATTEC、AUTAC以及AUTOTAC。
-
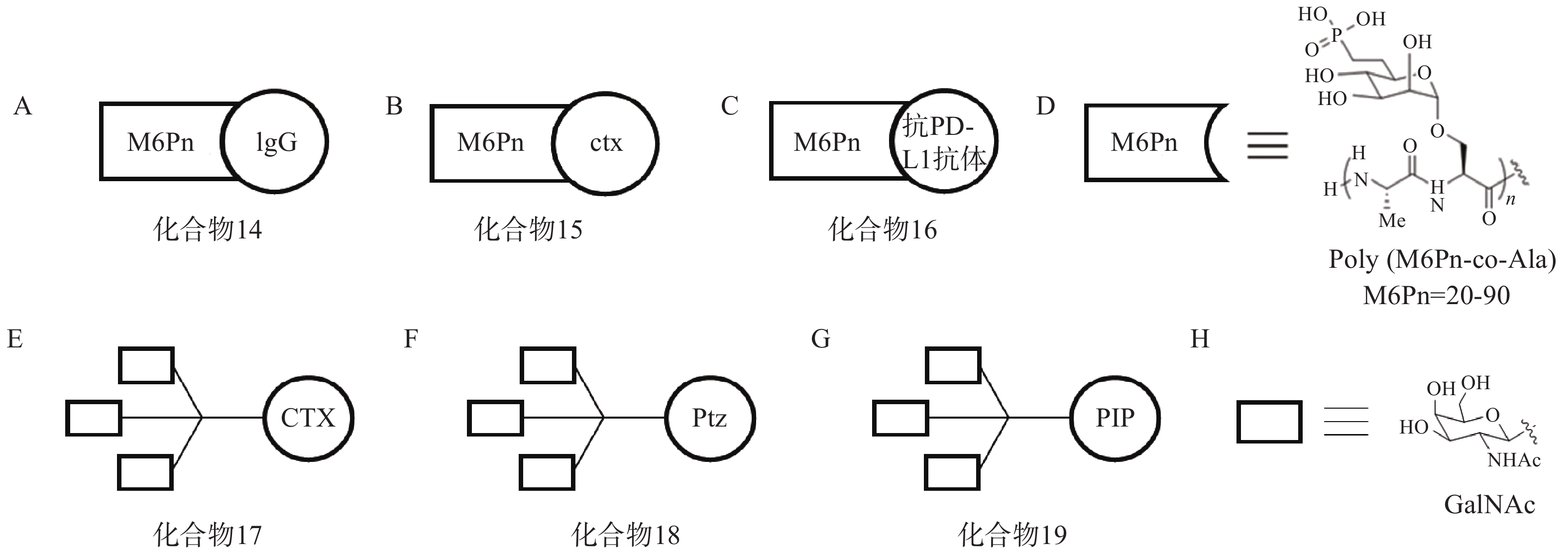
针对胞内蛋白的干扰和调控已经有相对成熟的研究和应用,但有约40%的基因编码产物为胞外及膜相关蛋白,例如细胞因子、趋化因子等,它们与肿瘤发生,衰老相关疾病以及自身免疫病等息息相关[63-64]。选择性降解该类蛋白具有改善人类健康的前景,但目前仍缺乏有力手段。2020年,斯坦福大学Bertozzi课题组首次提出LYTAC的概念[65],通过将小分子或抗体与低聚M6Pn糖肽配体相连,得到LYTAC分子。其作用机制如下:首先,低聚M6Pn糖肽配体与细胞表面溶酶体靶向受体CI-M6PR结合,另一端的小分子或抗体与靶蛋白特异性结合,在细胞膜上形成“CI-M6PR-LYTAC-靶蛋白”三元复合物;随后,CI-M6PR介导细胞膜凹陷,形成囊泡结构并包裹该复合物,转运至溶酶体中使底物降解,而CI-M6PR可返回细胞膜循环利用。
根据上述原理,该研究选择临床相关的靶点作为研究对象,验证LYTAC技术的可行性。以载脂蛋白E4(ApoE4)为例,研究人员将糖肽配体与多克隆抗小鼠IgG偶联形成LYTAC分子(化合物14,图6A)。生物实验结果显示,与单独使用小鼠ApoeE4一抗相比,化合物14使K562细胞中ApoE4-647的摄取增加了13倍,且在孵育后1 h和24 h均可观察到荧光信号与溶酶体的共定位。研究人员还将表皮生长因子受体(epidermal growth factor receptor,EGFR)阻断抗体西妥昔单抗(cetuximab,ctx)与糖肽配体偶联得到靶向EGFR的LYTAC分子(化合物15,图6B)。生物实验表明,10 nmol/L化合物15与Hela细胞孵育24 h后可诱导胞内超过70%的EGFR降解。此外,研究人员将Jurkat细胞与小鼠抗铁转蛋白受体-1(CD71)抗体和化合物14共孵育24 h后观察到高于80%的CD71降解;将抗程序性死亡配体1(PD-L1)抗体与糖肽配体相连形成LYTAC分子(化合物16,图6C),并用其处理PD-L1阳性的MDA-MB-231细胞,观察到细胞表面PD-L1水平降低了33%。
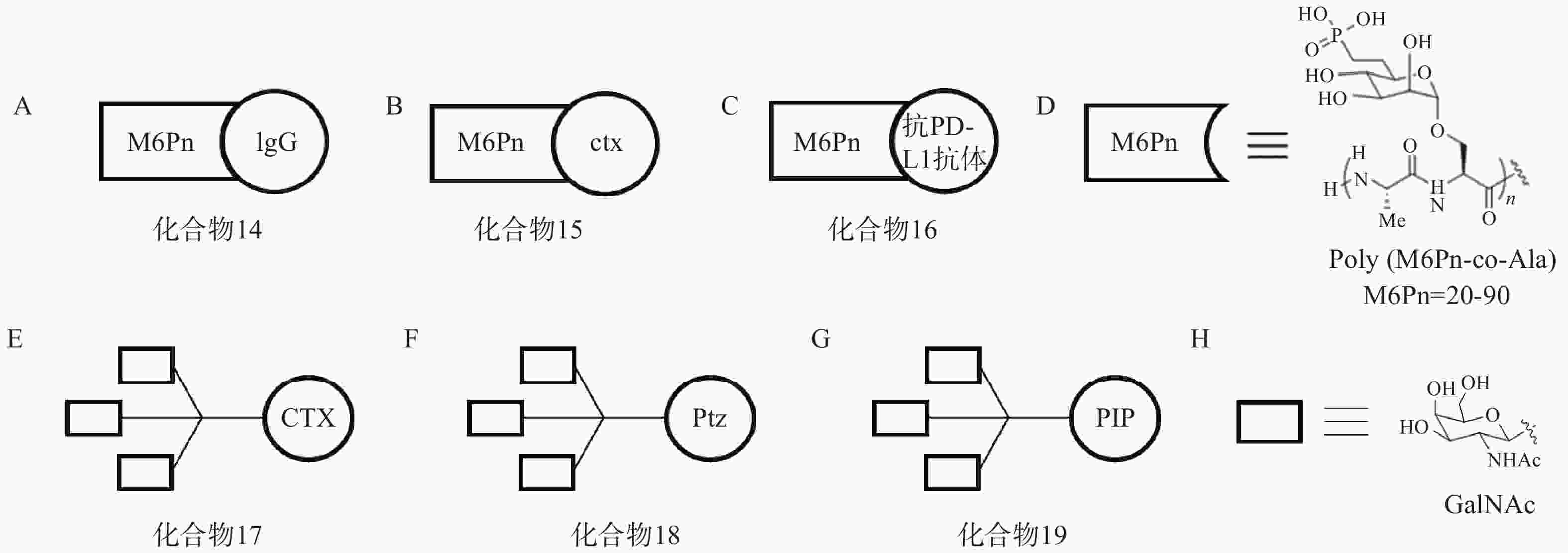
2021年,Bertozzi课题组再次基于LYTAC策略开发出多种具有肝组织特异性的LYTAC分子,并成功靶向降解肝癌细胞中的EGFR、HER2以及多种整合素蛋白[66]。去唾液酸糖蛋白受体(ASGPR)是一种在肝细胞膜特异性表达的溶酶体靶向受体,而三元N-乙酰半乳糖胺(tri-GalNAc)被证明是一种高效ASGPR配体。研究人员首先将ctx连接至tri-GalNAc基团,得到靶向EGFR的LYTAC分子(化合物17,图6E),Western Blot实验表明,化合物17在10 nmol/L下成功降解HEP3B细胞70%以上的表面EGFR蛋白,且在HeLa-GFP(ASGPR−,EGFR+,M6PR+)细胞中并未观察到EGFR的下调,证实了化合物17的肝组织靶向特异性;接着将HER2抗体pertuzumab(Ptz)连接至tri-GalNAc基团,得到靶向HER2的LYTAC分子(化合物18,图6F),实验表明,该化合物在100 nmol/L浓度下成功降解HEPG2细胞中75%的HER2蛋白,而单独的Ptz作用仅能下调30% HER2含量。
此外,该课题组还将多聚特异性整合素结合肽(PIP)连接至tri-GalNAc基团,得到靶向整合素家族蛋白的LYTAC分子(化合物19,图6G),生物实验表明,化合物19在100 nmol/L下成功降解HEPG2细胞中60%的整合素蛋白。
总之,LYTAC策略首次通过溶酶体途径选择性降解胞外及膜蛋白,为该类蛋白的调控提供了有价值的工具[67]。
-
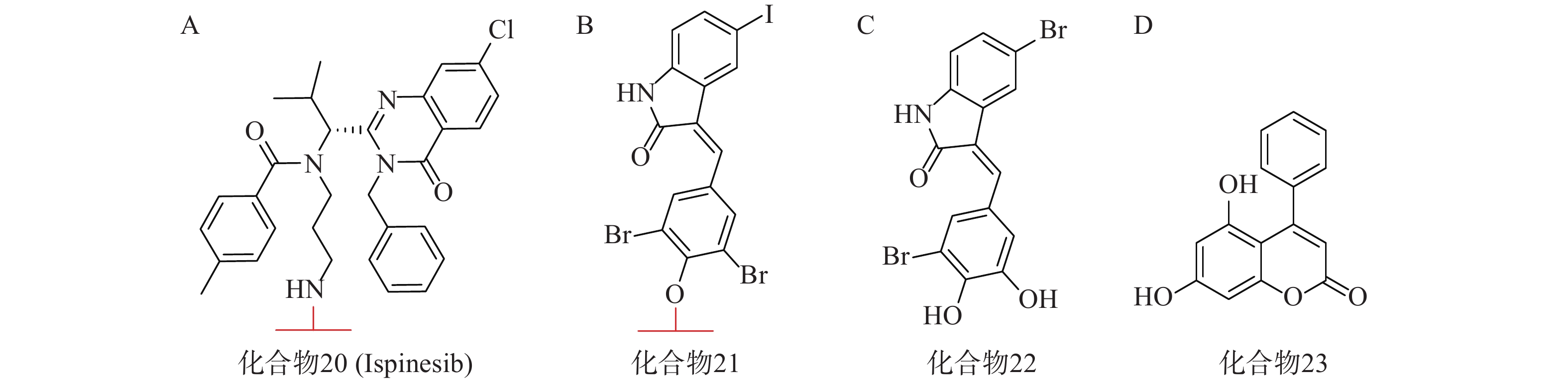
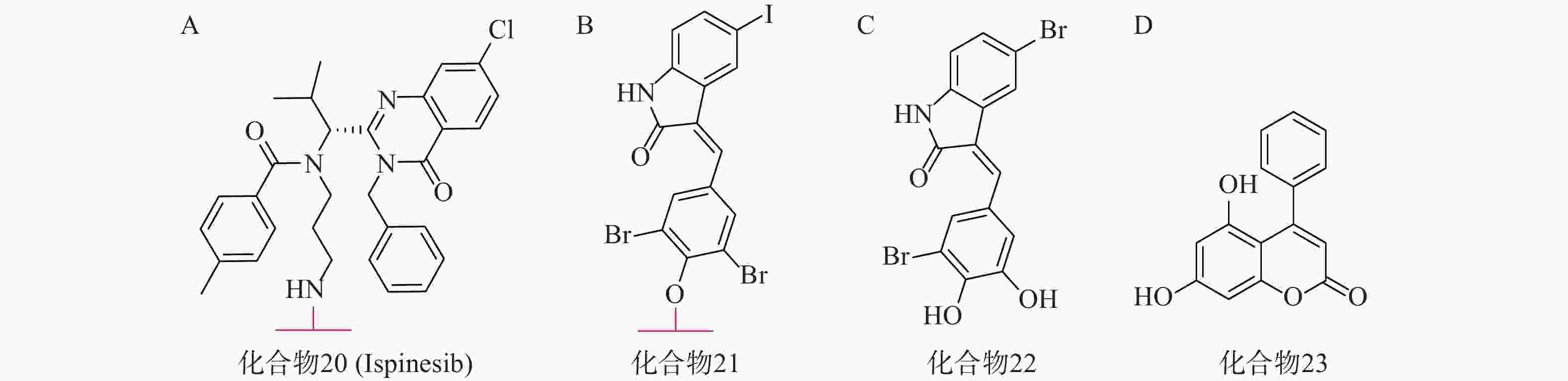
亨廷顿舞蹈症(Huntington’s disease,HD)是一种目前无法治愈的神经退行性疾病,其主要发病机理为致病突变亨廷顿蛋白(mHTT)在细胞内聚集。mHTT相比野生型亨廷顿蛋白(wtHTT)在结构上拥有一段多余的谷氨酰胺重复序列[68](PolyQ)。2019年,复旦大学鲁伯埙课题组在Nature发表研究成果,并首次提出ATTEC技术[69]。其作用机理类似于分子胶,能够同时结合LC3蛋白与mHTT,驱动mHTT进入自噬小体进行随后的降解。研究人员首先通过基于小分子微阵列(SMM)的高通量筛选,得到两个ATTEC小分子,即化合物20(KSP小分子抑制剂ispinesib,图7A)和化合物21(图7B)。结构分析发现,它们均含有一个芳环,并连接到卤素取代的芳基内酰胺双环。接着基于类似物筛选又得到另外两个ATTEC小分子,即化合物22(图7C)和化合物23(图7D)。这四种分子能够特异性识别mHTT中的PolyQ,而不与wtHTT相互作用。体外实验表明,这些ATTEC分子均能在50~100 nmol/L的浓度范围下有效降解HD小鼠原代皮层神经元中的mHTT,且不影响wtHTT。研究人员进一步考察了这些化合物在HD转基因果蝇和小鼠体内模型中的影响,证明了这些ATTEC分子能明显下调果蝇体内以及小鼠大脑皮层中mHTT的含量,并改善疾病相关的表型。该研究开创性地提出了一种新的靶向蛋白降解策略,首次使用小分子化合物参与自噬途径实现选择性降解,为polyQ相关疾病提供了有前景的治疗手段。
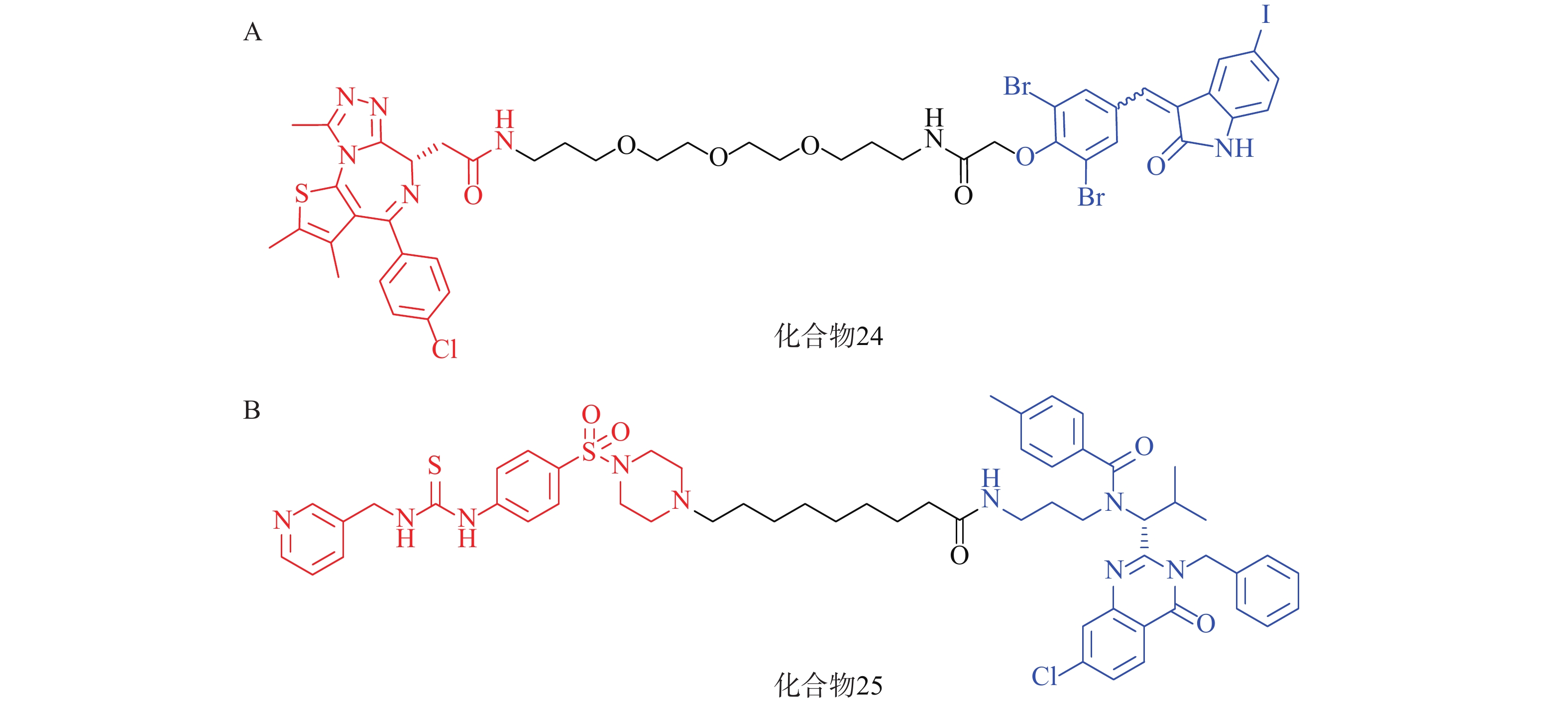
2021年,四川大学欧阳亮课题组基于ATTEC设计思路,以化合物 21为LC3蛋白配体,BET经典抑制剂JQ1为BRD4配体,两者通过不同类型和长度的Linker相连,合成了一系列靶向BRD4的自噬降解剂。其中活性最优的化合物24(图8A)在多种癌细胞中均能诱导BRD4的有效降解,并发挥抗增殖作用。该研究成功利用ATTEC策略拓展了经典靶点BRD4的降解途径[70]。
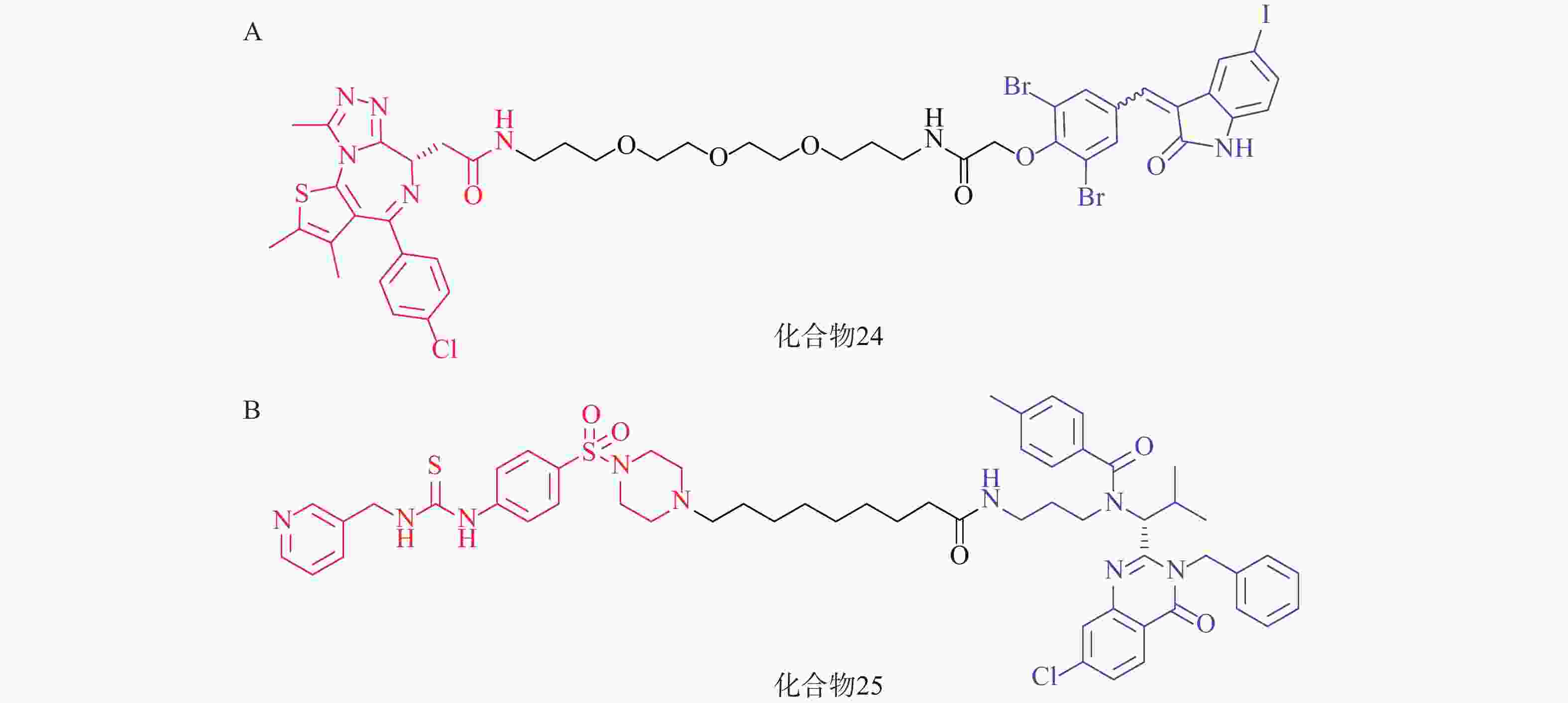
基于相似的策略,笔者所在的课题组以ispinesib(化合物20,图7A)为LC3配体, NAMPT抑制剂MS2为靶蛋白配体,设计了一系列靶向烟酰胺磷酸核糖转移酶(NAMPT)的自噬降解剂。其中,活性最优的化合物25(图8B)在100 nmol/L浓度下可降解A2780细胞中90%的NAMPT,且化合物25对于A2780细胞的抑制活性(IC50=46 nmol/L)显著高于NAMPT抑制剂MS2(IC50=490 nmol/L),具有进一步研究的价值[71]。
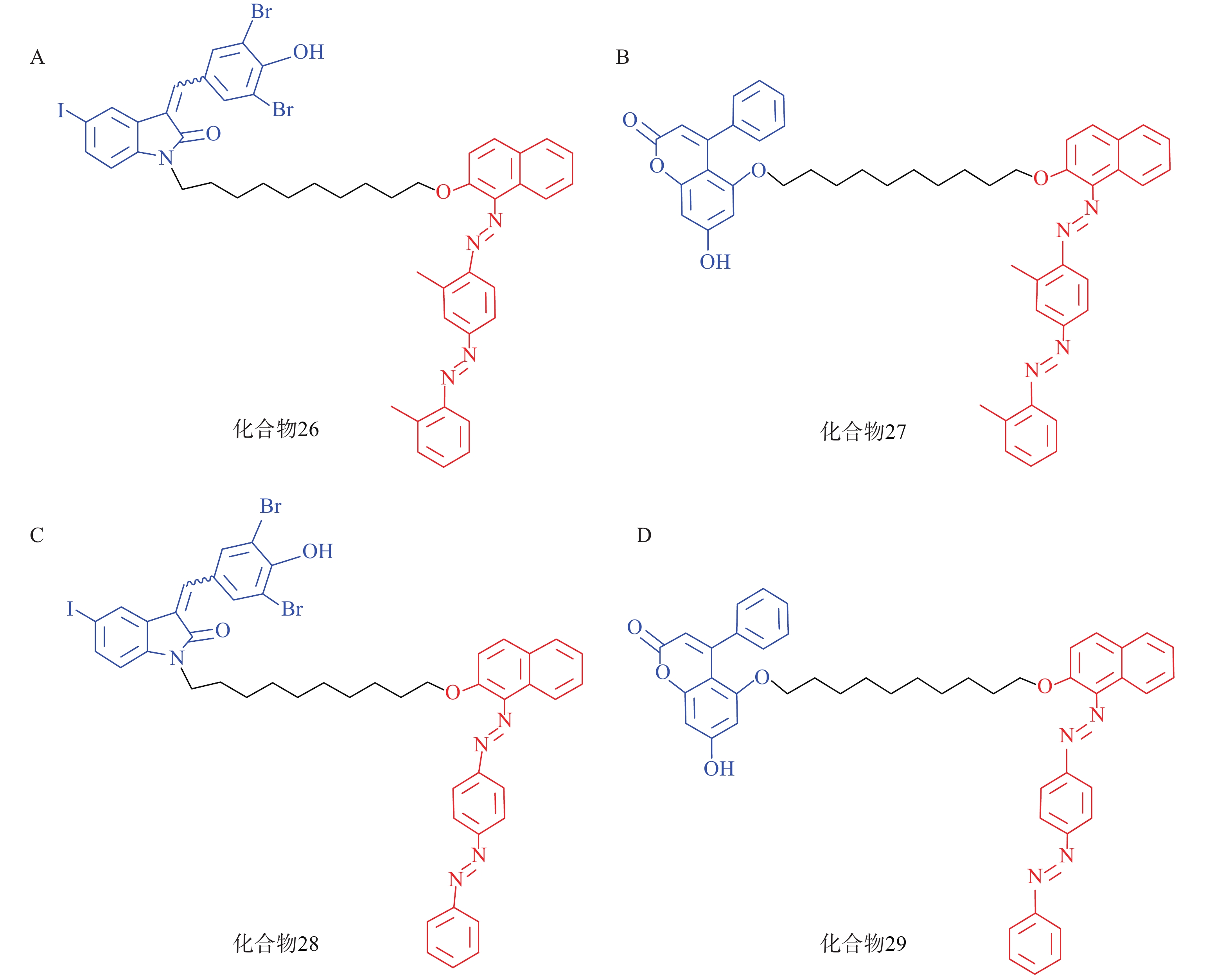
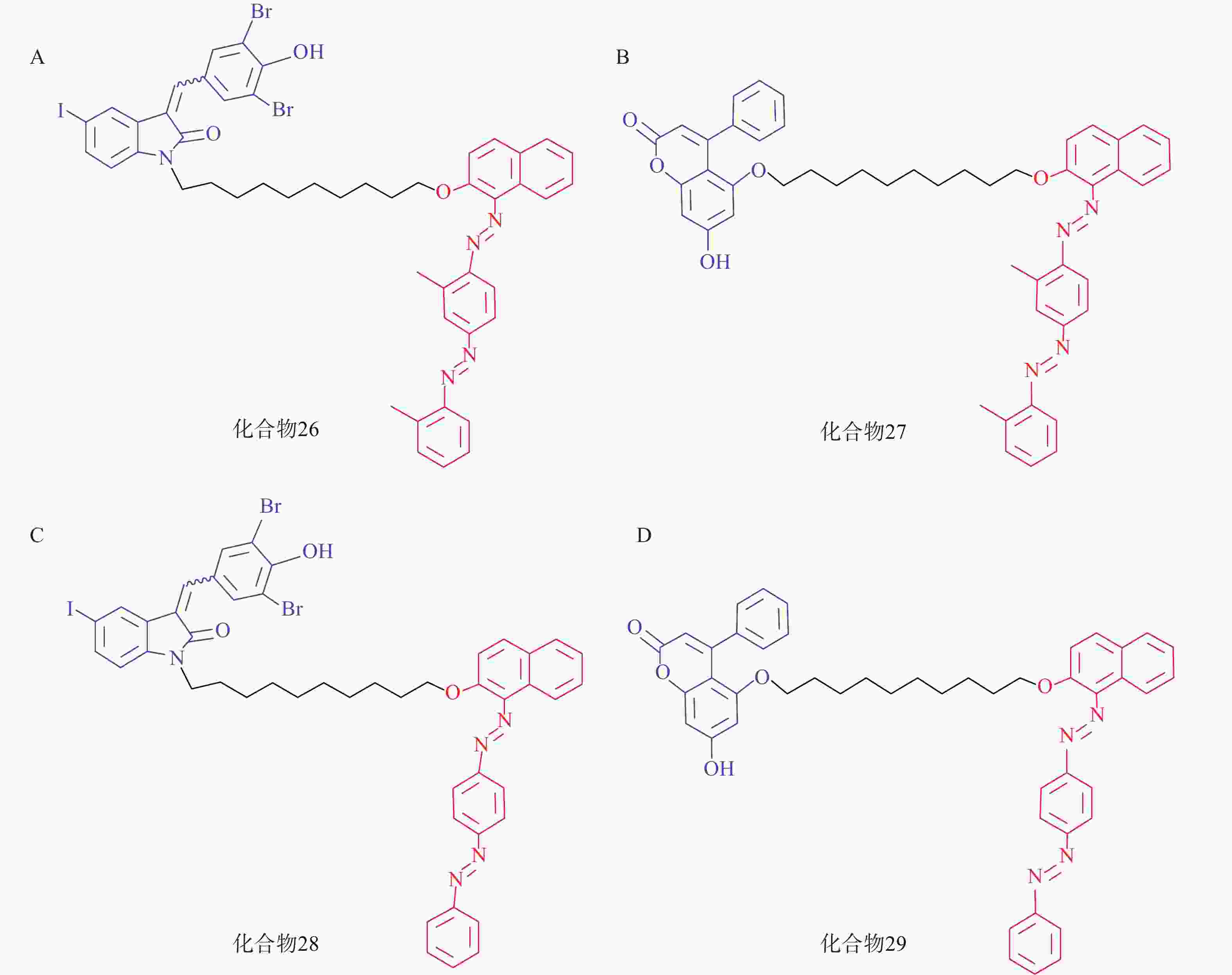
脂肪液滴(LD)是普遍存在于细胞中的储脂结构,通过磷脂单分子膜将其中的脂类物质(如三酰甘油等)包裹。LD的异常积聚与多种疾病有关,如脂肪肝、神经退行性病变以及心血管疾病等。2021年,鲁伯埙课题组基于ATTEC策略设计了4种靶向LD的小分子化合物(化合物26~29,图9),它们以化合物21或化合物23作为LC3配体,通过Linker与LD配体苏丹Ⅲ或苏丹Ⅳ相连。生物实验表明,所有化合物在5 μmol/L下均能显著降低脂肪细胞内的LD含量。此外,通过腹腔注射化合物28和化合物29(30 mg/kg,每天1次),肥胖症小鼠模型(db/db小鼠,C57BL/6J - Leprdb/Leprdb)的体重在2周内显著降低约15%;给药结束后处死小鼠并取出肝脏,观察到小鼠血清和肝脏中的三酰甘油和总胆固醇含量显著降低。在CDAHFD诱导的非酒精性脂肪肝(NASH)小鼠模型的体内实验中也观察到与上述一致的结论。细胞内非蛋白致病底物一直以来都是极具挑战性的课题,该研究首次利用ATTEC策略靶向清除非蛋白物质并改善相关表型,丰富了靶向降解的应用范围[72]。
-
K63泛素化修饰是一种靶向自噬-溶酶体途径的特异信号,即底物蛋白与泛素K63位连接后,可被细胞内固有的自噬受体(p62/SQSTM1,简称p62)通过泛素相关结合域(UBA)选择性识别,p62再通过特定活性位点与LC3蛋白结合,进而诱导底物进入自噬小体,最后与溶酶体融合完成降解[73]。2019年,Arimoto课题组在一项关于抗菌自噬的研究中得到启发,证明了S-鸟苷化(S-guanylation)可以介导底物的K63泛素化和选择性自噬清除,并基于此提出AUTAC技术[74]。AUTAC由3部分构成:靶蛋白配体,作为降解标签的S-鸟嘌呤衍生物,以及将两者相连的Linker。该类分子通过靶蛋白配体与底物结合,降解标签进一步诱导靶蛋白发生K63泛素化,再被p62识别并基于ALP降解。
该研究共设计了4种AUTAC分子(化合物30~33,图10),其中化合物30~32分别靶向MetAP2、FKBP12和BRD4蛋白。Western blot结果显示,化合物30在1~100 μmol/L的浓度梯度下均可显著下调Hela细胞内MetAP2的水平;化合物 31在10 μmol/L浓度下能有效降解Hela细胞内FKBP12蛋白;值得注意的是,化合物32以相同方式处理Hela细胞后,在不同浓度下仅表现出微弱的降解作用,这可能与BRD4蛋白的核定位有关。
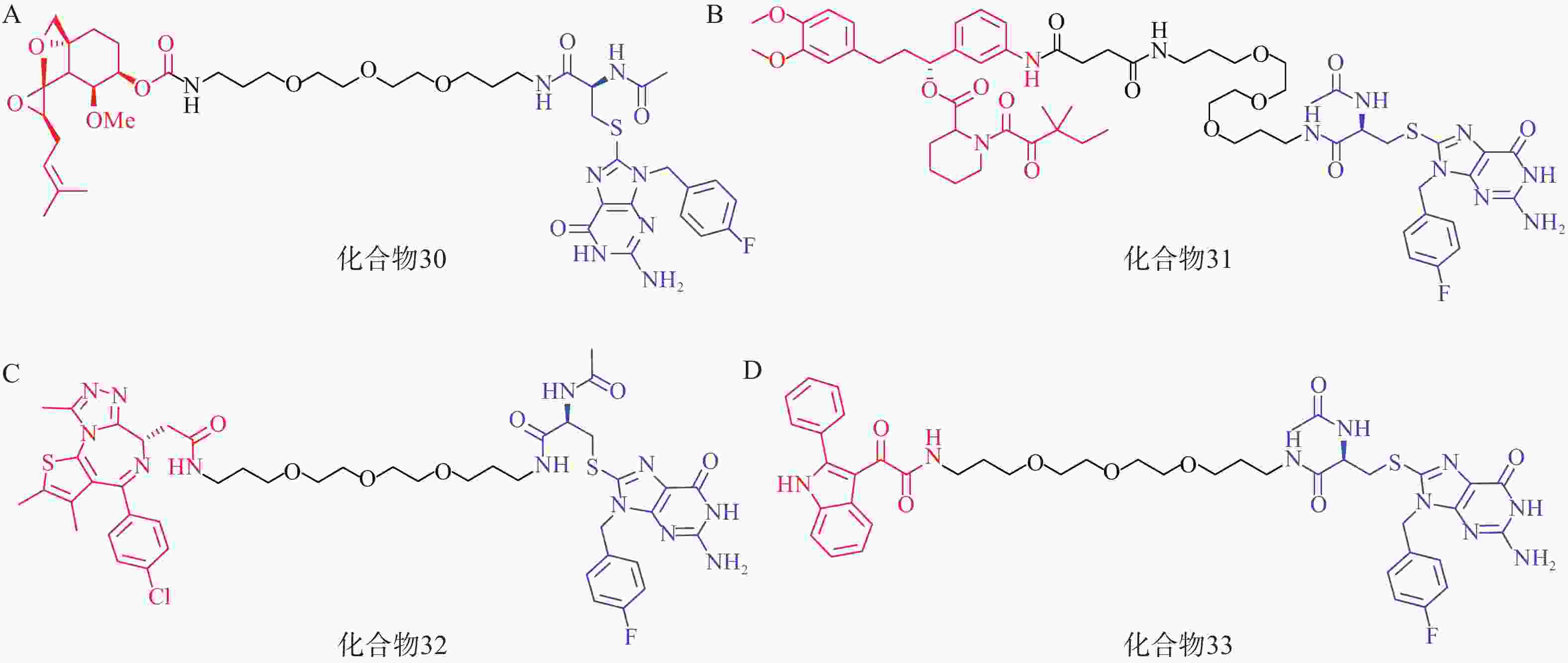
此外,AUTAC技术还可以用于靶向降解线粒体碎片等细胞器。唐氏综合征(DS)是一种遗传疾病,线粒体功能障碍是其核心致病因素。DS患者成纤维细胞(Detroit 532细胞)中线粒体形态比野生型成纤维细胞更加碎片化,且部分膜电位丢失。利用化合物33(10 μmol/L)处理Detroit 532细胞,可在24~72 h内诱导细胞内线粒体自噬,并观察到细胞线粒体形态和膜电位显著改善。
该研究为靶向蛋白降解领域增添了新的化学工具,给线粒体功能损伤相关疾病的治疗提供了新的思路。但该手段依赖泛素化过程,目前已报道的相关应用较少,普适性仍有待验证,且S-鸟嘌呤衍生物诱导K63泛素化的机制仍有待阐明,降解标签的类药性优化也颇具挑战性。
-
2022年,Yong课题组首次提出AUTOTAC技术[75]。AUTOTAC是一种异双功能分子,一端为p62配体,另一端为靶蛋白配体,两者通过Linker相连。具体作用机制如下:AUTOTAC与p62和靶蛋白同时结合,形成三元复合物,导致p62构象发生变化,产生自身寡聚并暴露出LC3(light-chain 3)结合域,进而与 LC3的结合,介导底物隔离至自噬小体并通过溶酶体途径降解。
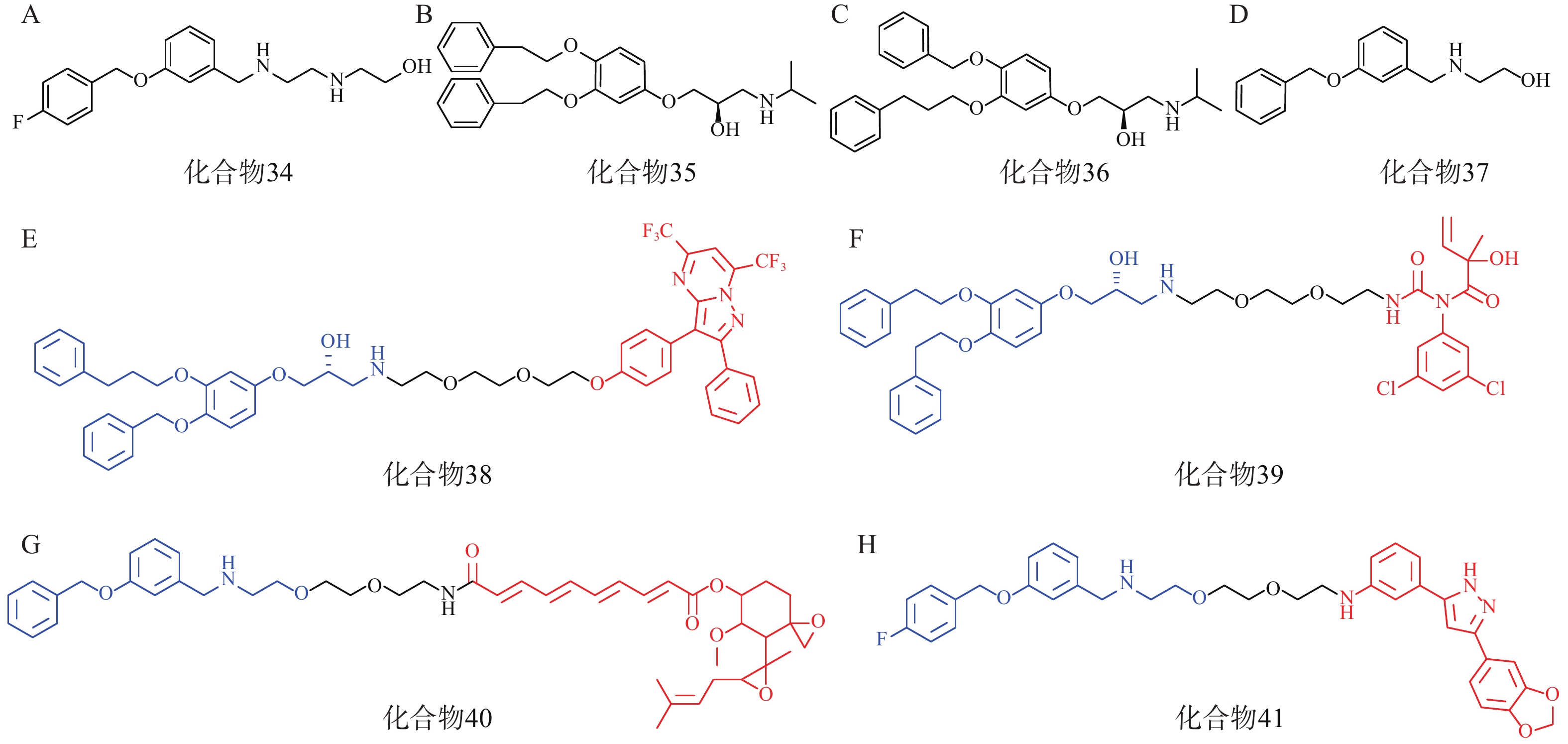
研究人员首先筛选并优化得到了4个拥有p62高亲和力的分子:化合物34~37(图11A、11B、11C、11D)。接着,以这些分子的片段为p62配体,设计合成了多个AUTOTAC分子,即化合物38~40(图11E、11F、11G),分别用于靶向降解ERβ、AR和MetAP2。生物实验表明,化合物38能在HEK293T细胞中基于自噬溶酶体途径高效降解ERβ(DC50=1.48 nmol/L);靶向AR的化合物39和靶向MetAP2的AUTOTAC 40分别能有效降解LNCaP前列腺癌细胞中的AR(DC50=211.08 nmol/L)或HEK293T细胞中的MetAP2(DC50 ≈ 0.70 μmol/L)。
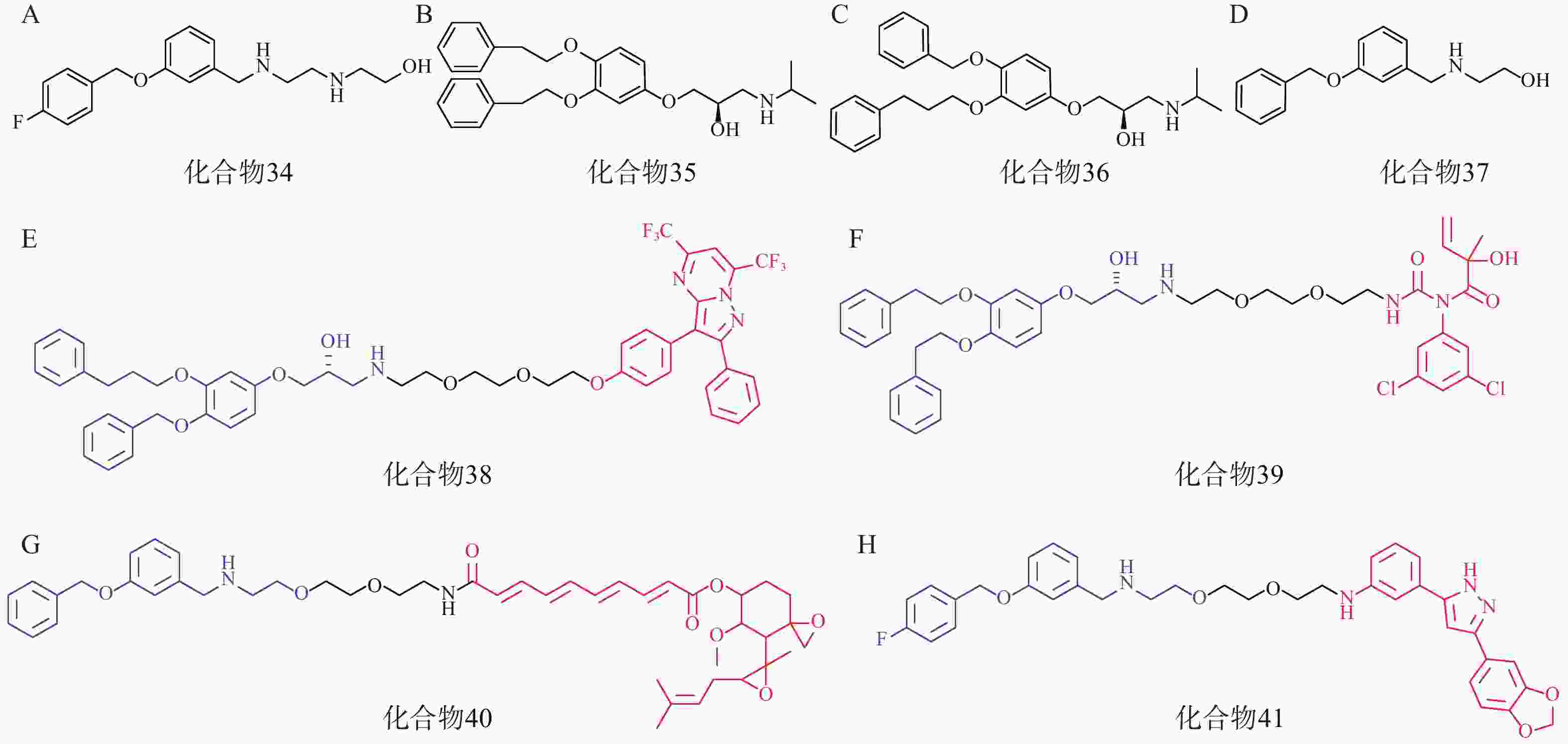
此外,在神经退行性病变等蛋白质病中,错误折叠的蛋白质容易形成聚集体。Anle138b是一种处于临床研究阶段的可选择性结合蛋白聚集体的小分子药物,研究人员将p62配体与Anle138b通过聚乙二醇链相连得到了靶向蛋白聚集体的AUTOTAC分子(化合物41,图11H),其在10 nmol/L下能高效降解SHSY5Y细胞中的P301L tau突变体(DC50=3 nmol/L)。
该研究首次通过靶向自噬受体实现了对多种单体蛋白及致病蛋白聚集体的清除,不依赖泛素化过程,为基础研究和药物开发提供了一项选择性自噬降解蛋白的技术。
-
靶向蛋白降解技术利用真核细胞内固有的两大蛋白降解系统——泛素-蛋白酶体系统(UPS)和溶酶体途径,实现了多种疾病相关蛋白的靶向降解,正引领着下一代小分子药物的发展方向。目前已经有多个PROTAC分子或分子胶在临床试验中取得了积极的结果,但大部分的靶向蛋白降解技术尚处于研究的早期阶段,并面临着诸多挑战。例如:①大部分异双功能降解剂分子量较大,相比传统药物透膜性和口服生物利用度较差,难以实现临床转化;②某些基于多肽和寡核苷酸的降解剂面临着储存、递送和体内代谢稳定性的难题;③多数蛋白降解技术的机制研究尚不完善,例如LC3配体与蛋白如何作用、S-鸟苷化如何诱导底物K63泛素化等问题仍有待研究;④基于溶酶体途径的降解策略普遍活性有限,活性提升难度较大,已报道的体内研究较少,普适性有待验证。
近年来,研究人员围绕上述问题积极探索,开发了多维度的靶向蛋白降解技术,例如:①基于点击化学提出的CLIPTAC以小分子形式被细胞摄取,进入细胞后通过点击反应形成活性PROTAC分子发挥作用,有望提高药物的水溶性及细胞渗透性[76];②靶向递送策略,包括抗体偶联蛋白降解剂[77]、光控PROTAC(如pc-PROTAC[78]和photoPROTAC[79])等对降低毒副作用,提高药物靶向性具有深远意义。下一阶段,科研人员一方面需要不断开发新的降解技术,挑战更多“难成药”靶点,另一方面,应持续优化现有降解技术,包括扩展E3连接酶的选择并进行合理的PROTAC设计,完善选择性自噬降解机制的研究,提高蛋白降解剂的成药性并实现临床转化。
Advances and prospects in targeted protein degradation
doi: 10.12206/j.issn.2097-2024.202303008
- Received Date: 2023-03-06
- Rev Recd Date: 2023-05-11
- Publish Date: 2023-06-25
-
Key words:
- targeted protein degradation /
- ubiquitin–proteasome system /
- lysosomal degradation pathway /
- drug discovery /
- PROTAC
Abstract: Targeted protein degradation (TPD) techniques eliminate pathogenic proteins by hijacking the intracellular proteolysis machinery which includes the ubiquitin-proteasome system (UPS) and the lysosomal degradation pathway, holding promise to overcome the limitations of traditional inhibitors and further broaden the target space including many “undruggable” targets, and provide new targeted treatments for drug discovery. In this review, recent advances in a variety of promising TPD strategies were summarized, such as proteolysis targeting chimera (PROTAC), molecular glue, lysosome-targeting chimaera (LYTAC), autophagosome-tethering compound (ATTEC), autophagy-targeting chimera AUTAC and AUTOTAC, particularly. The representative case studies, potential applications and challenges were analyzed.
| Citation: | ZHOU Luozhu, SHENG Chunquan. Advances and prospects in targeted protein degradation[J]. Journal of Pharmaceutical Practice and Service, 2023, 41(6): 341-351, 365. doi: 10.12206/j.issn.2097-2024.202303008

|
 DownLoad:
DownLoad: