

-
老药新用是药物设计的重要途径之一,采用此方法可以获得药物研究所需的先导化合物,迄今为止已有许多成功的案例,特别是在新型冠状病毒肺炎治疗药物研究中采用此策略,获得了很多有价值的药物[1-3]。阿司匹林从发明至今已有百年的历史,最初用于解热、镇痛和抗炎。随着医学科学的发展,在临床上发现阿司匹林的新用途,能够抑制血小板的聚集,抑制心脑血管疾病的发生[4-5]。因此,从上市药物中寻找新的适应证或将其作为先导物进行结构优化的老药新用设计方法成为药物研究的有效手段[6-7]。
HTML
-
实验所用试剂均为分析纯;6种药物原料药:苄普地尔、保泰松、阿洛西林、盐酸氮䓬斯汀、盐酸多沙普伦、美芬洛酮(南京多点化工有限公司);还原型辅酶Ⅱ(NADPH,东京化学工业有限公司);混合人肝微粒体(美国BD Gentest公司)。
Biotek Synergy H2多功能酶标仪、MK-2全自动酶标仪、戴安公司Ultimate3000液相色谱系统(包括三元输液泵、柱温箱、自动进样器和真空脱气机)和AB公司4000Q-Trap型串联质谱仪,配有电喷雾电离源,以及AB公司Analyst (version 1.5.1)、LightSight (version 2.2.1)数据采集及分析软件。
-
将荧光肽(PMDM-F,Anaspec)用缓冲溶液(100 mmol/L磷酸钾、0.02%叠氮化钠、100 μg/ml牛丙种球蛋白,用蒸馏水定容至500 ml,pH 7.5)稀释至10 nmol/L,加入到梯度稀释的MDM2癌基因蛋白中,30ºC避光孵育30 min。荧光各向异性值用Biotek Synergy H2多功能酶标仪读取,蛋白结合常数根据荧光各向异性值用Mathematica 9软件拟合得到。
将购买的6个药物溶于二甲基亚砜(DMSO),缓冲液稀释至所需要浓度(最终缓冲液中含有1%DMSO)。然后将20 μl化合物加入到60 μl含有10 nmol/L PMDM-F肽和100 nmol/L MDM2蛋白的溶液中,30ºC避光孵育1 h。荧光各向异性值用Biotek Synergy H2多功能酶标仪读取,蛋白结合常数根据荧光各向异性值用Mathematica 9软件拟合得到。
-
将人肺腺癌上皮细胞(A549)培养,待细胞生长至70%~80%时,加药处理。药物处理不同时间后,用0.25%胰酶消化,离心收集细胞,再用胞浆蛋白核蛋白裂解液4 ℃裂解,提取胞浆蛋白及核蛋白。采用12 %SDS-PAGE凝胶电泳,将蛋白转移到固相支持膜(PVDF膜)上,封闭液室温封闭1~2 h,用等渗缓冲盐溶液(tris-buffered saline tween-20, TBST)洗膜5 min,洗3次。采用TBST稀释抗体,4 ℃孵育过夜,TBST洗膜5 min,洗3次。然后用TBST稀释HRP标记的二抗,室温孵育1 h,TBST洗膜5 min,洗3次。暗室内采用ECL显色,X线片曝光成像。以甘油醛-3-磷酸脱氢酶(GAPDH)为内参照,分析成像光片灰度值,并以对照组为基准进行量化。
-
样品配制:用DMSO溶解后,加入磷酸缓冲盐溶液(PBS)配成1000 μg/ml的溶液或均匀的混悬液,然后用含DMSO的PBS稀释。
MTT比色法:96孔板每孔加入浓度为(5~6)×104个/ml的细胞悬液100 μl,置37ºC,5% CO2培养箱内。24 h后,加入样品液,10 μl/孔,设双复孔,37ºC、5% CO2作用72 h。每孔加入5 mg/ml的MTT溶液20 μl,作用4 h后加入溶解液,100 μl/孔,置培养箱内,溶解后用MK-2全自动酶标仪测定570 nm处A值,计算IC值。
IC(%) =[(空白对照孔A值-给药孔A值) /空白对照孔A值]×100%
根据不同浓度的IC值,进行线性回归,计算出抑制肿瘤细胞生长50%的药物浓度,即IC50。
-
每个孵育体系总体积为200 µl,介质为100 mmol/L磷酸缓冲液(PBS,pH7.4),包括终浓度为1 mg/ml的肝微粒体蛋白、50 µmol/L的苄普地尔和2 mmol/L的还原型辅酶Ⅱ(NADPH),用37°C水浴进行孵育,60 min后加入同体积冰冷乙腈终止反应。被终止反应的肝微粒体孵化样品,离心5 min(13000 r/min),取出全部上清液置于10 ml试管中,于40ºC空气流下吹干,残留物以100 μl乙腈/水(10: 90, V/V)复溶,取10 μl进行HPLC/Q-Trap MS分析。色谱条件为色谱柱为Kromasil® C18(150 mm×2.1 mm,5 μm),Agilent C18保护柱(12.5 mm ×2.1 mm,5 μm)。流动相:乙腈(A)–10 mmol/L甲酸铵水溶液(B),梯度洗脱:0~2 min,10% A;10~15 min,90% A;15.1~18 min,10% A;流速为0.3 ml/min;柱温为室温;进样量为10 μl。质谱条件为电喷雾离子化源(ESI),正离子检测。雾化温度为500 ℃,电喷雾电压为5200 V,气帘气流速20 L/min,雾化气50 L/min,辅助气流速50 L/min。扫描方式为MRM\MIM簇发EPI。
2.1. p53-MDM2蛋白结合抑制活性测试[8]
2.2. 蛋白印迹试验[9]
2.3. 体外抗肿瘤活性测试[10]
2.4. 体外代谢研究[11]
-
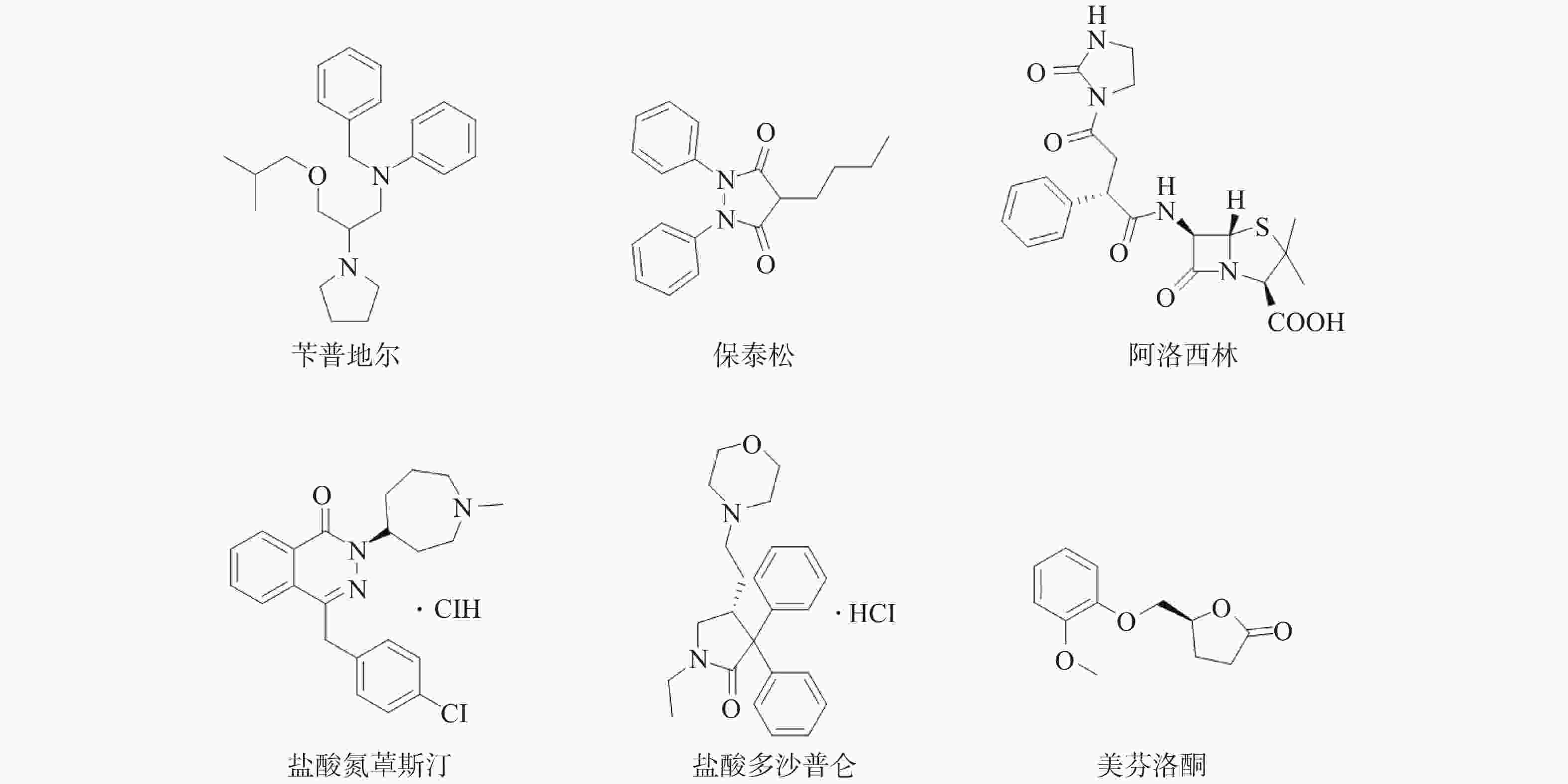
课题组在针对p53-MDM2靶标进行虚拟筛选过程中,发现Wayne等已运用计算机构象筛选法对ZINC数据库中3244个FDA已批准上市药物进行了筛选,鉴别出与p53-MDM2抑制剂Nutlin-3a结构不同,但具有类似形状电荷分布的化合物[12]。然后,通过Autodock软件与MDM2蛋白进行分子对接,根据结合能获得打分函数排名前15的化合物,但未用分子药理学实验证实其作用靶标。因此,课题组购买了可售的6个药物(图1)。对这6个药物进行蛋白结合抑制活性实验,以Nutlin-3为阳性对照药,结果见表1。
化合物 Ki
(μmol/L)IC50
(μmol/L)U-2OS
(wt-p53)Saos-2
(p53 null)A549
(wt-p53)NCI-H1299
(p53 null)Nutlin-3 0.093 12.2 8.38 2.18 1.97 美芬诺酮 5.79 >100 >100 8.43 >100 盐酸氮䓬斯汀 216.3 6.38 3.80 1.54 4.32 苄普地尔 0.456 2.58 1.56 1.04 1.39 保泰松 155.5 71.31 >100 91.45 >100 阿洛西林 NA >100 >100 >100 >100 盐酸多沙普仑 NA >100 >100 60.68 >100 从表1中可以看出,6个药物中美芬诺酮、盐酸氮䓬斯汀、苄普地尔和保泰松4个药物体现出一定的p53-MDM2蛋白结合抑制活性,其余2个药物无活性。其中,钙离子拮抗剂苄普地尔的Ki值达到0.456 μmol/L,显示出优异的p53-MDM2蛋白结合抑制活性。
为验证苄普地尔是否能抑制p53-MDM2蛋白结合,采用免疫印迹试验测定相关蛋白的表达变化,结果如图2。从图中可以看出,苄普地尔对p53蛋白表达作用相对较小,并且随着苄普地尔浓度上升到10 μmol/L,反而出现下降。但苄普地尔能显著降低MDM2蛋白的表达,而且呈剂量依赖关系,初步说明苄普地尔能明显抑制p53-MDM2蛋白结合。

-
课题组选择骨肉瘤(U-2OS、Saos-2)和肺癌(A549、NCI-H1299) 2组细胞株进行了6个药物的体外抗肿瘤活性测试(表1)。从表1中可以看出,苄普地尔对4种细胞株均具有优秀的抗肿瘤活性,其IC50值均低于3 μmol/L,优于阳性对照药Nutlin-3。而美芬诺酮尽管具有中等的p53-MDM2蛋白结合抑制活性(Ki=5.79 μmol/L),但除对A549具有较好的抗肿瘤活性,其余细胞株均未显示出活性。令人意外的是盐酸氮䓬斯汀尽管具有较弱的p53-MDM2蛋白结合抑制活性,但对4种肿瘤细胞株也显示出较好的活性。
-
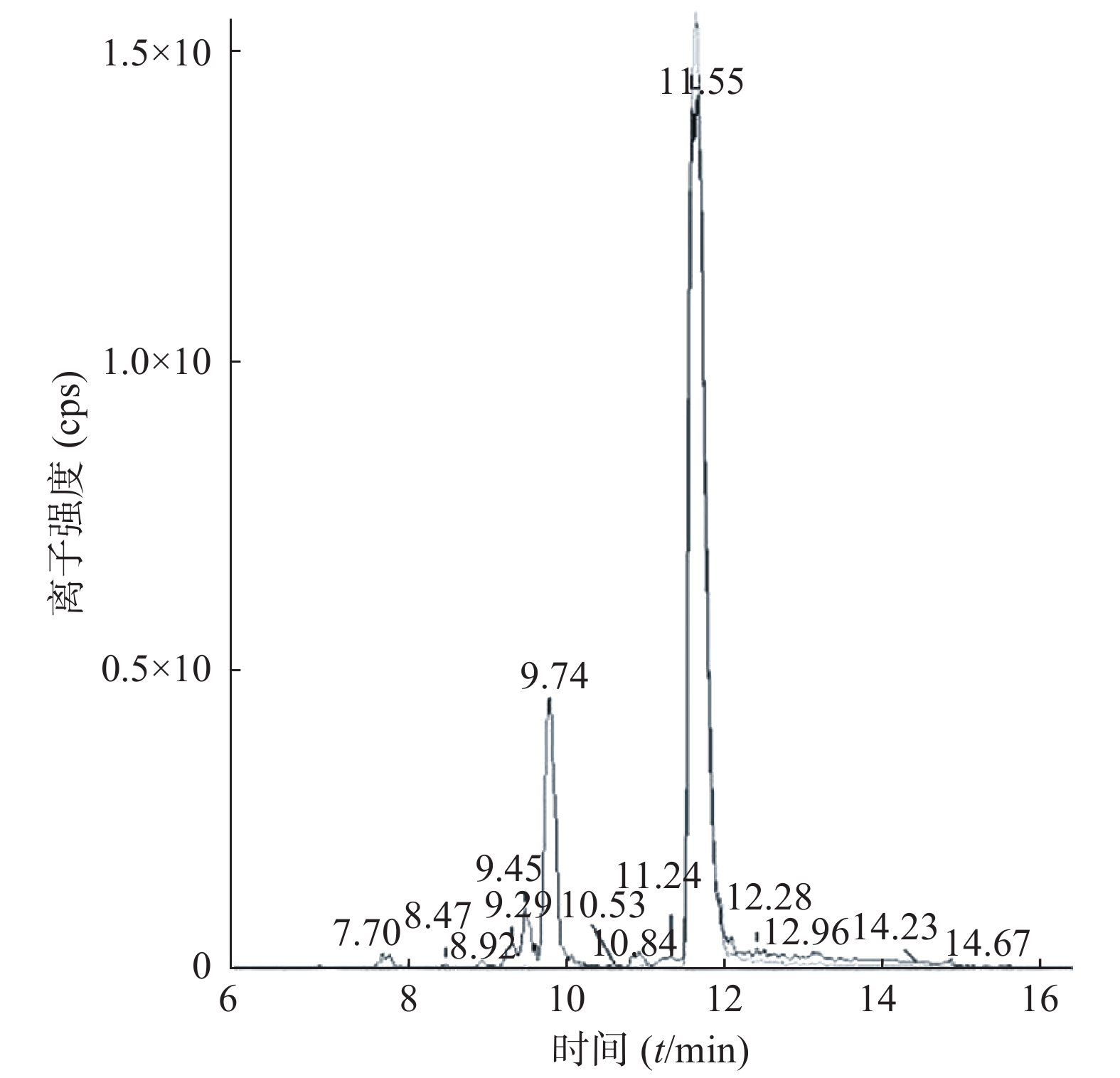
课题组进一步对苄普地尔开展了人肝微粒体中代谢产物研究,结果见表2、图3和图4。发现在经人肝微粒孵化后的样品中除原形药物(M0,m/z 367)外,在保留时间为9.8 min处检测到1个色谱峰,命名为M1,其m/z为383。推测其分子式组成为C24H34N2O2,比原形增加1个O,可能为单氧化代谢产物。在MS/MS扫描质谱图中,由M1获得的主要碎片离子为m/z 312、212、184(图4),其中m/z 312、212比原形的主要碎片离子m/z 296、196,相对分子质量增加16,而碎片184与原形的相同,且未见相对分子质量少18的碎片,推测其为苯环羟基化代谢产物。
编号 代谢途径 质荷比 分子式 保留时间(t/min) M0 原形 367 C24H34N2O 11.6 M1 氧化 383 C24H34N2O2 9.8 
3.1. p53-MDM2蛋白相互作用抑制活性
3.2. 体外抗肿瘤活性研究
3.3. 苄普地尔的体外代谢研究
-
基于老药新用的药物设计思想,课题组验证发现钙离子拮抗剂苄普地尔具有优秀的抗肿瘤活性和较强的p53-MDM2蛋白结合抑制活性。进一步通过免疫印迹试验发现,苄普地尔能显著降低MDM2蛋白的表达,而且呈剂量依赖。体外代谢研究发现,苄普地尔在人肝微粒体中的代谢产物主要是苯环羟基化单氧化代谢产物。研究结果表明,苄普地尔可作为p53-MDM2蛋白结合小分子抑制剂先导化合物,用于后续的结构优化设计研究。

 DownLoad:
DownLoad: